最近闲暇时间,对自己的富含bug包做了简单的维护(实话说,我现在很少遇到bug了,感觉修的七千八八了,但肯定还是不完善的)。
前言
主要是fixbatch做了下更新:
- 添加了指定内参基因的功能
housekeep_gene,例如指定actinB,GAPDH等 - 添加了`
edgeR的利用count值,数据矫正后用CPM做定量分析的功能edger_fix,以期能解决批次效应 - 增加
pvca,计算各批次的影响大小 - 增加
combat_fix,根据sva包copy而来,对已知的批次效应进行矫正
附带着也更新了xbox的功能
get_df不管输入数据框是data.frame,tibble,还是matrix都支持转换成data.frame格式get_tb不管输入数据框是data.frame,tibble,还是matrix都支持转换成tibble格式- 增加
dat2matrix功能,将data.frame或者tibble格式转成matrix格式(其实我应该下次更新的时候,改成get_mt会统一,我感觉)
也完善了autopca的一些报错机制
但是这都是一些小的修补。最近主要是在学习sva的功能和使用方法。后面有时间的话,就将sva的功能整理到fixbatch中, 做个整合和补充,有点大杂烩的感觉。
后面有时间的话,我会用pkgdown完善下说明文档。目前还是没时间去摸索pkgdown.
devtools::update_packages("fixbatch")
devtools::update_packages("xbox")
devtools::update_packages("autopca")sva 学习笔记
言归正穿,因为最近一直在看sva的说明文档,以及Bioconductor的Gene Expression Normalization Workflow
真的当自己去看这些东西的时候,才更清晰的认识到,之前fixbatch包中根据中位数矫正的norm_fix以及根据稳定基因计算矫正因子矫正的edger_fix.其实都应该算是一种数据标准化的,类似于scale,rlog乃至vst,就是一种将数据压缩再同一单位的量程内,并保留数据大小的相对关系的处理。
但自己真去看sva的时候,才发现,其是将整个实验的变量分为adjustment variables和variables of interest,我简称为待修正变量(非关注变量)和关注变量。其中关注变量是实验设计中关注的差异效应(CK-VS-T),而非关注变量就是其他无关的,但是在实验过程引入的变量,例如实验的批次,试剂的批次,人员的批次,测序的批次,甚至包括材料本身的批次(背景噪音)。最极端的应该是医学上的研究。科研中是无法控制人的遗传背景,生活习性,以及饮食习惯,甚至年龄,性别有时候都无法选择。这些极大的个体之间的差异,会显著影响我们所关注的变量效应,所以真实的数据结果中包含的应该是,两组因素都含有的。
image-20201223230258676
这就导致了我们在进行数据分析之前,要尽量从实验设计上避免此类非关注变量的影响。那么如果实验中真的有此类的非关注因素影响。又该怎么办呢。
首先针对不同的情况要做如下区分:
- 是否已知有哪些非关注因素
- 未知的非关注因素
已知非关注因素
假设我们已经知道了批次效应,那我们可以用PVCA(Bushel, 2013)来估计基因表达数据的主成分(PCA)的各个因素的方差比例。将高纬数据的总方差归因到每个给定因素的协变量。剩余未分配的方差分数被认为为“剩余”方差(“residual” variance)。
PVCA有效地结合了主分量分析 (PCA)的算法, 减少了高纬数据的维度。同时通过方差分量分析 (VCA), 将因素当作随机效应进行混合线性模型,来估计其所占到总方差的比例。所解释的方差是按每个因素对 PC 的贡献的加权平均值计算的,您可以选择通过设置 PC 解释的方差阈值来指定要包含的各要素数量。
当然我也是一知半解的!0.o,看了不一定懂,懂了也说不出来,要不然为啥写学习笔记。写出来有时候就会自己反复思考。
总之,我知道这个PVCA可以得到各个非关注因素的方差贡献度,这就足够了,我当然想看整个实验设计中,有哪些因素影响,以及各个因素影响的大小。
PVCA本身是个Bioconductor 包,其输入数据必须是要biobase格式的。biobase我不熟悉。而且我不喜欢Bioconductor的R包,其复杂的输出格式,提示信息以及依赖关系(谁用谁知道)。
后面发现Gihub上有前辈已经改过一次,可以用来直接适配咱们常用的数据框格式的。但是我下载用了以后发现,这尼玛,不行不行的。步步报错(考虑不全),各种依赖包不注释(非充分检查R包的依赖关系),
实话说,这水平还不一定比我强呢!
所以我还是将其改改,收录在我自己的R包fixbatch中吧,这个能算抄袭吗?毕竟我在原来的基础上,改的bug的也不少了。

image-20201224141212846
pvca听起来很复杂,其实简单举个例子,就会知道其多么有用了。我们使用bladderEset数据集来做这个分析。这是一个包含了57个样本,5个批次的膀胱的芯片表达量数据,是biobase类型的数据集合。其实我们就用到两个其中,一个是数据表达矩阵(row:gene/feature, col:sample),一个批次效应矩阵 (row:sample, col:factor)。
# BiocManager::install("bladderdata")
# pcva需要最新版本的fixbatch,没有更新的需要更新下
# devtools::update_packages("fixbatch")
library(bladderbatch)## 载入需要的程辑包:Biobase## 载入需要的程辑包:BiocGenerics## 载入需要的程辑包:parallel##
## 载入程辑包:'BiocGenerics'## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, basename, cbind, colnames,
## dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
## grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
## rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
## union, unique, unsplit, which, which.max, which.min## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.library(fixbatch)
library(sva)## 载入需要的程辑包:mgcv## 载入需要的程辑包:nlme## This is mgcv 1.8-31. For overview type 'help("mgcv-package")'.## 载入需要的程辑包:genefilter## 载入需要的程辑包:BiocParallellibrary(limma)##
## 载入程辑包:'limma'## The following object is masked from 'package:BiocGenerics':
##
## plotMAdata(bladderdata)
# 提取批次效应矩阵
pheno = pData(bladderEset)
# 提取表达矩阵
edata = exprs(bladderEset)
# analysis
pvcaObj <- pvca(edata, pheno[,-1], threshold = 0.7,inter = FALSE)
# plot
pvca_plot(pvcaObj)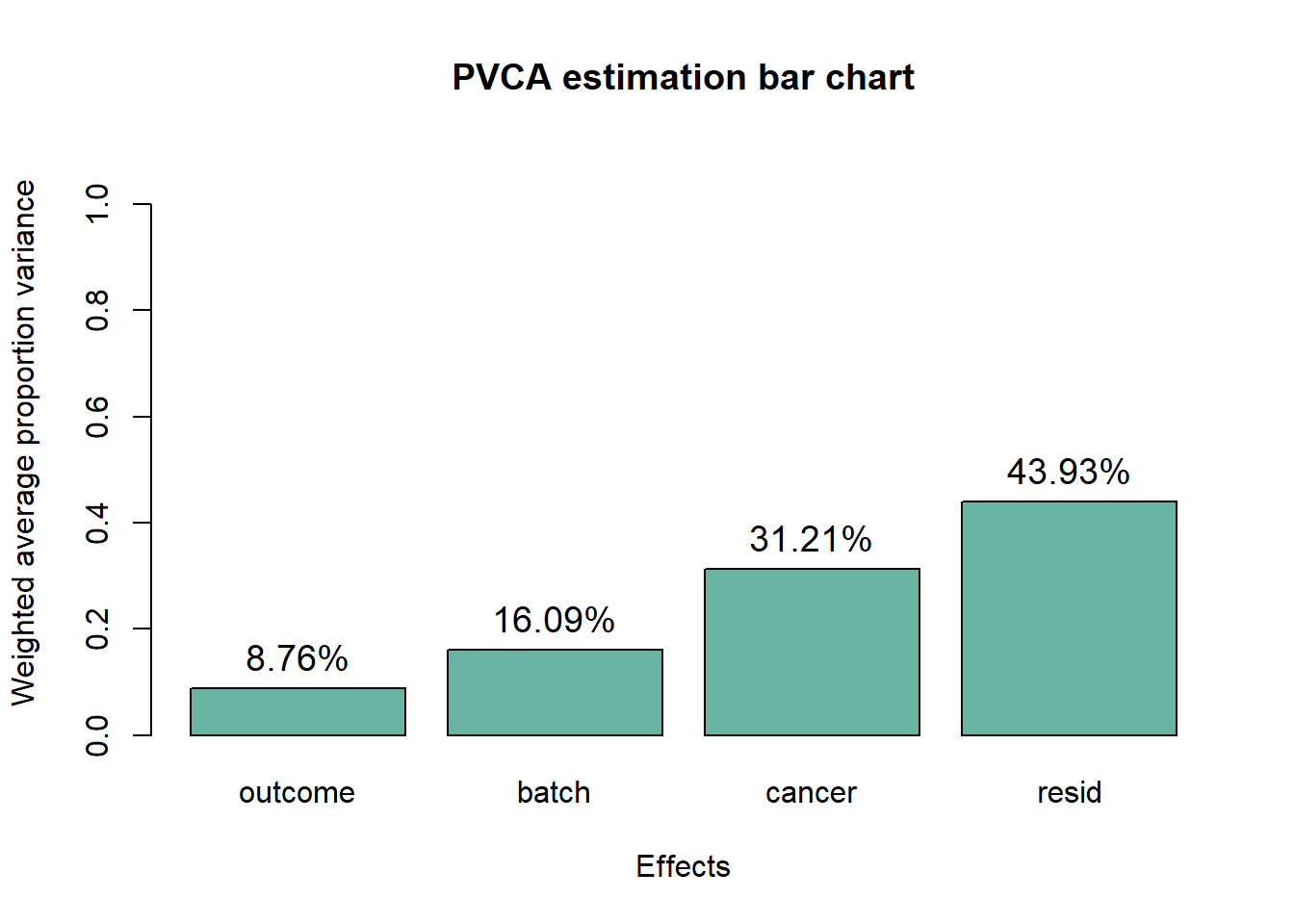
我们可以看到其批次效应 (非关注因素)outcome, batch,总共加起来,占比才不到30%。而关注因素cancer才占比31%,而还有43%的解释度,还是未知的。
| outcome | batch | cancer | resid |
|---|---|---|---|
| 0.087601 | 0.1609 | 0.3121 | 0.439 |
那么如果我们假设我们找不到其他的未知变量了,必须要从以上两个变量中选个,我们肯定选比例最大的batch, 因此我们就会用到combat来进行“已知非关注因素”的矫正。
# 首先先创建一个包含需要矫正变量的矫正矩阵,但是我们关注的batch不用加进来,因为这是另一个参数的输入,因为没有需要矫正的变量。所以这是一个截距1的矩阵
modcombat = model.matrix(~1, data=pheno)
# 输入需要矫正的批次,只能一个变量,多了报错,后续会更新功能支持多个
batch = pheno$batch
# 开始矫正
combat_edata = ComBat(dat=edata, batch=batch, mod=modcombat)## Found5batches## Adjusting for0covariate(s) or covariate level(s)## Standardizing Data across genes## Fitting L/S model and finding priors## Finding parametric adjustments## Adjusting the Datacombat 最大的有点是是会返回一个矫正后的表达矩阵。这点就可以方便进行后续的PCA和热图的绘制。combat是sva的功能,但是我把它简单包装下,放在我的fixbatch中,命名为combat_fix。
未知非关注因素(copy sva users guide)
刚刚上面,我们是假设我们已知所有的非关注因素了,但其实还有至少50%左右的方差比,我们没有找到对应的因素。所以在这里可以利用sva的功能来寻找** surrogate variables(替代变量)**。
开始之前,我们先用设计两个模型矩阵,一个是包含所有因素(包括关注因素和非关注因素),另一个是只有非关注因素的模型矩阵。
# all variables
mod = model.matrix(~as.factor(cancer), data=pheno)
# adjustment variables(no variables need to djusting)
mod0 = model.matrix(~1,data=pheno)有了这两种模型矩阵,我们就可以去探究一些未知的非关注因素了。首先要确定目标的替代变量的数目,然后再计算其值。
# caculate the number of the surrogate variables
n.sv = num.sv(edata,mod,method="leek")
# get the value of surrogate variables on every sample
svobj = sva(edata,mod,mod0,n.sv=n.sv)## Number of significant surrogate variables is: 2
## Iteration (out of 5 ):1 2 3 4 5结果中存储着预估替代变量的值,然后与最开始的两个模型矩阵进行合并,得到更新后的模型矩阵。
modSv = cbind(mod,svobj$sv)
mod0Sv = cbind(mod0,svobj$sv)至此,已经完成替代变量的计算和合并,现在利用已有的模型矩阵,用limma进行差异分析,其步骤如下:
fit = lmFit(edata,modSv)
# 这个比较矩阵的设计是我最懵逼的地方,我现在还是不理解,但是这一步的目的是为了计算cancer-vs-normal的差异比较。
contrast.matrix <- cbind("C1"=c(-1,1,0,rep(0,svobj$n.sv)),"C2"=c(0,-1,1,rep(0,svobj$n.sv)),"C3"=c(-1,0,1,rep(0,svobj$n.sv)))
# 得到差异分析的模型
fitContrasts = contrasts.fit(fit,contrast.matrix)
# 得到差异结果
eb = eBayes(fitContrasts)
topTableF(eb, adjust="BH")## C1 C2 C3 AveExpr F P.Value
## 207783_x_at -13.45607 0.26592268 -13.19015 12.938786 8622.529 1.207531e-69
## 201492_s_at -13.27594 0.15357702 -13.12236 13.336090 8605.649 1.274450e-69
## 208834_x_at -12.76411 0.06134018 -12.70277 13.160201 6939.501 4.749368e-67
## 212869_x_at -13.77957 0.26008165 -13.51948 13.452076 6593.346 1.939773e-66
## 212284_x_at -13.59977 0.29135767 -13.30841 13.070844 5495.716 2.893287e-64
## 208825_x_at -12.70979 0.08250821 -12.62728 13.108072 5414.741 4.350100e-64
## 211445_x_at -10.15890 -0.06633356 -10.22523 9.853817 5256.114 9.845076e-64
## 213084_x_at -12.59345 0.03015520 -12.56329 13.046529 4790.107 1.260201e-62
## 201429_s_at -13.33686 0.28358293 -13.05328 12.941208 4464.995 8.675221e-62
## 214327_x_at -12.60146 0.20934783 -12.39211 11.832607 4312.087 2.257025e-61
## adj.P.Val
## 207783_x_at 1.419929e-65
## 201492_s_at 1.419929e-65
## 208834_x_at 3.527673e-63
## 212869_x_at 1.080599e-62
## 212284_x_at 1.289423e-60
## 208825_x_at 1.615555e-60
## 211445_x_at 3.133969e-60
## 213084_x_at 3.510132e-59
## 201429_s_at 2.147888e-58
## 214327_x_at 5.029329e-58我摊牌了,这一趴,关于替代变量这部分,我是根据说明文档整理来的。但是这个比较组的设计我很懵逼,希望各位有大佬能给我解惑下,不胜感激!
已知其变量顺序为: outcome batch cancer sv1 sv2
而contrast.matrix内容如下:
print(contrast.matrix)## C1 C2 C3
## [1,] -1 0 -1
## [2,] 1 -1 0
## [3,] 0 1 1
## [4,] 0 0 0
## [5,] 0 0 0那为啥contrast.matrix代表的是去掉批次效应后的cancer-vs-normal的差异比较呢。
后续我找到答案,我也会及时更新的。
总结下:目前fixbatch也算小有成就,不仅包含了norm_fix,edger_fix,以及combat_fix这些批次矫正的脚本,还有pvca这种探究因素效应的神器。在批次矫正中,应该是足够用了,功能暂时不想更新了。后续就是完善说明文档和补BUG吧。