遇到项目问题了
事情是这样的,遇到了一个项目,在审核的时候。发现结果有强的批次效应。在样品重复性分析中基本上没有任何规律,差异基因也少。
至于差异基因那些事,可以看我昨天的文章为什么差异基因少, 差异到底是什么?
虽然后面也找了原因。但是原因只是一种双方扯皮时候的依据。问题还是要回到实际项目中,如何矫正这种批次效应。这就不得不开动自己的脑瓜子想想怎么矫正批次效应。
看看什么是情况
具体项目是什么情况呢,我们一起来看看,当然了,数据是不能透漏的。我这边自己造了个假数据。用来做案例的重新。
注意:本文中不仅用了我自己创建的R包autopca来画PCA图,还用了另一个自己的R包xbox中的数据格式转换功能。甚至还新建了我的第三个R包fixbatch。顾名思义,这个新R包目标是为了矫正批次效应。目标是很大的,但现实中批次效应这个范围太大,我第一能力不足,很难写个新算法,第二很难超越别人的R包。所以这个R包还是于己方便,做个大杂烩,以已有的R包做主题,然后再按照自己的思路和想法,做些修鞋匠的工作,就是些胶粘磨补而已。
自己写的文章,用自己的R包,不算硬广吧!毕竟咱的R包也没人用,自己再不用,那就真的不仅垃圾,而且无用了。╮(╯_╰)╭
首先是加载R包,如果没有安装,请自行安装
# BiocManager::install("edgeR")
# BiocManager::install("preprocessCore")
# devtools::install_github("wangjiaxuan666/autopca")
# devtools::install_github("wangjiaxuan666/xbox")
# devtools::install_github("wangjiaxuan666/fixbatch")
# install.packages("patchwork")如果那位大佬之前已经安装我的R包autopcaor xbox。那可就惨了,一定要记得更新,因为我自己R包富含BUG。我之前每次使用自己的R包写脚本,都是写脚本10min,改自己的BUG至少3小时起。
反而用自己的R包更浪费时间呢,难顶!
所以如果用了我富含BUG的R包,一定要及时更新!一定要及时更新!一定要及时更新!重要的事情说三遍。
就像这样,每次使用的时候,请用如下代码,选择Github来源更新方式。
#devtools::update_packages("autopca")
#devtools::update_packages("xbox")
#devtools::update_packages("fixbatch")目前为止,R包已经全部更新和安装了,那就开始正文。批次效应是啥样,以及如何矫正。
require(autopca)
require(xbox)
require(fixbatch)
require(tidyverse)
require(edgeR)
require(preprocessCore)
require(patchwork)
# read data
setwd(r"(D:\QQ_files\992914078\FileRecv)")
dat = readr::read_tsv("gene_exp_data.xls")
#用R包autopca中的pca_data_tidy函数整理数据,支持输入tibble或者dataframe格式数据
dat_pca_nofix = pca_data_tidy(dat)## ... Notice: the input data is a tibble
## ... Wonderful: the input data vaule in every column all be numberic value
## ... Successed! the pca data save in the object#用R包autopca中的pca函数画图
p1 <- pca(dat_pca_nofix,rename = "replace",
str_sample = "_fpkm",
str_group = "-\\d$",
display_sample = T,
add_ploy = T) +
ggtitle("Unnormalised dat")
print(p1)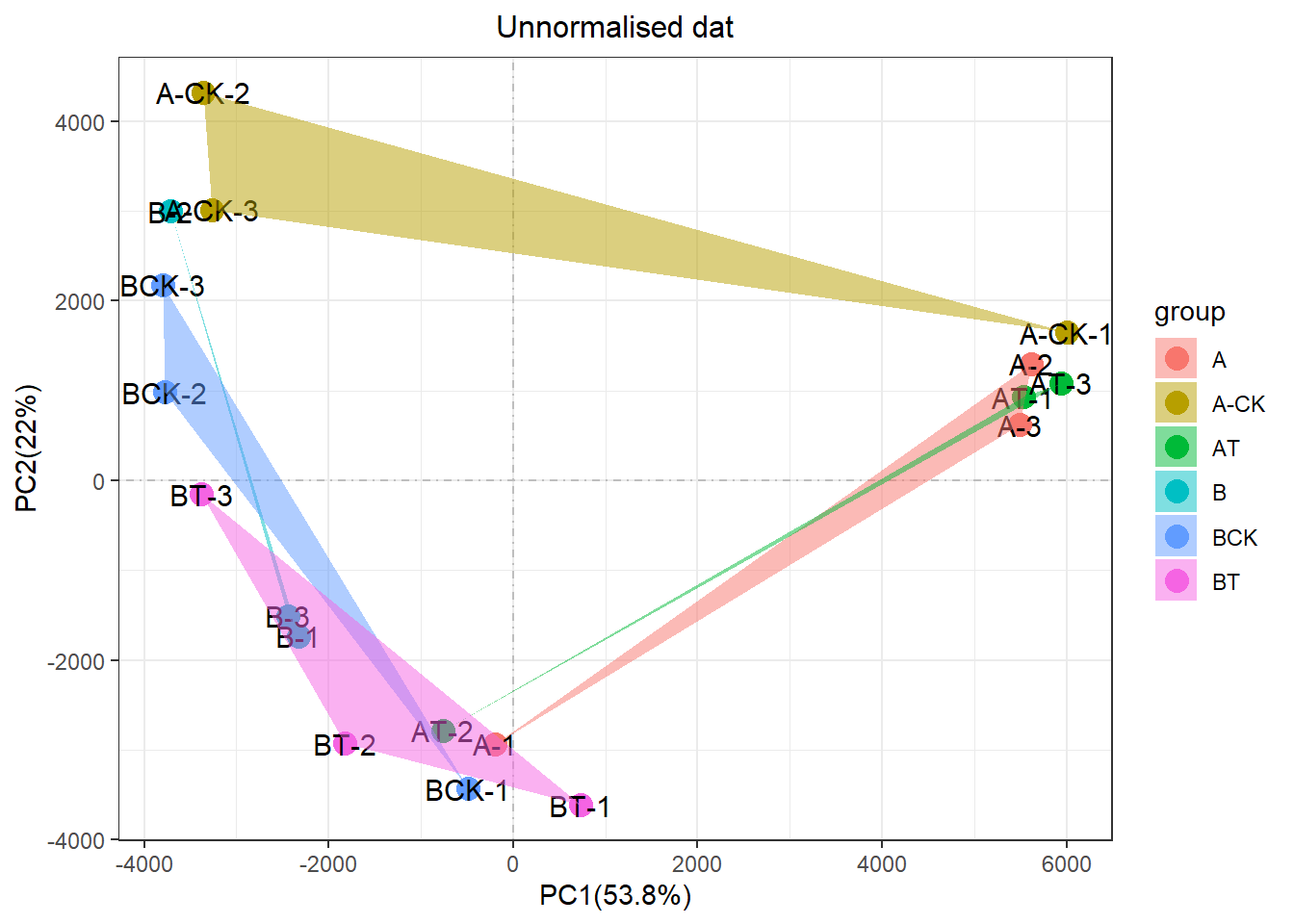 可以很明显的发现,这个一个非常差的重复性。同组的三个生物学重复,左边和右边都有,彼此撕裂。PC1占比40%多,但是这个因素却不是实验处理。而是一种“未知”神秘力量影响。
可以很明显的发现,这个一个非常差的重复性。同组的三个生物学重复,左边和右边都有,彼此撕裂。PC1占比40%多,但是这个因素却不是实验处理。而是一种“未知”神秘力量影响。
剧透下,这个批次效应已经被解决了,神秘力量也知道是什么,但具体的就不再剧透了,连续剧要一集一集看。 内心OS:我才不会说,是因为我没时间写文章
最开始呢,我发现离群样本的基因表达箱线图的上限要比正常样本高一些。所以我首先想到了根据中位数的方法进行批次效应。
没办法中位数矫正的方法,即是公司中口口相传的矫正方法,其实其形式和DeSeq2以及edgeR的矫正都有点异曲同工之妙。而且这个项目的箱线图确实很有规律。所以首先想到的是中位数矫正方法。
当然edgeR和DeSeq2的矫正方法考虑的东西更多一些。但是如果你在Youtube上看过statquest的视频,就会知道edgeR和DeSeq2也是根据“稳定基因”计算矫正因子。和中位数矫正的方法真的很想。我甚至用了edgeR的calcNormFactors函数计算了校正因子,并假设测序数据量一样的情况,用矫正因子矫正数据,和中位数矫正的结果非常类似。
中位数矫正的方法,主要用的是preprocessCore包,但是我利用这个R包,感觉效果不好,所以我想了这个问题。如果根据所有基因来进行中位数矫正批次效应,那肯定是不符合生物学规律的,因为肯定是有些基因就是发生很大的变化,如果这个变化的基因数目达到了一定的量级。那中位数矫正就不再是矫正了,而是矫正过枉了。而且所有基因都参与计算矫正,很难出现比较好的效果。
所以我根据edgeR和DeSeq2的思想,想着如果大部分基因都是不发生变化的,我就专门找其中“可靠的,稳定表达的基因”来计算矫正因子,然后再回到整体数据中进行矫正。
确定了这个思路,我就新建了四个函数,分别是计算变异系数的函数col_cv,以及计算基因表达量之和的col_sum以及寻找管家基因的housekeep,最后还有矫正数据的norm_fix。我将这四个函数打包成R包。其效果如下:
#构建数据 start-----------------------------
y <- matrix( rpois(1000, lambda=5), nrow=200 )
n = dim(y)
colnames(y) = paste(rep("sample",n[[2]]),1:n[[2]])
rownames(y) = paste(rep("gene",n[[1]]),1:n[[1]])
y = as.data.frame(y)
y[,1] <- (y[,1]*0.05)
y[,2] <- y[,2]*5
#构建好数据 end-----------------------------
#画图展示数据
col <- RColorBrewer::brewer.pal(n[[2]], "Paired")
par(mfrow=c(1,3))
##原始数据
boxplot(y, las=1, col=col, main="")
title(main="A. Example: Unnormalised data",ylab="Gene Exp")
##用preprocessCore函数矫正
y_fix1 = preprocessCore::normalize.quantiles(as.matrix(y))
boxplot(y_fix1, las=1, col=col, main="")
title(main="C. Example: Normalised data by normalize.quantiles",ylab="Gene Exp")
##用自建R包fixbatch中矫正
y_fix2 = norm_fix(y,expcv = 1, exphigh = 1, explow = 0)## The stable housekeep gene number is 198boxplot(y_fix2, las=1, col=col, main="")
title(main="C. Example: Normalised data by fixbatch",ylab="Gene Exp")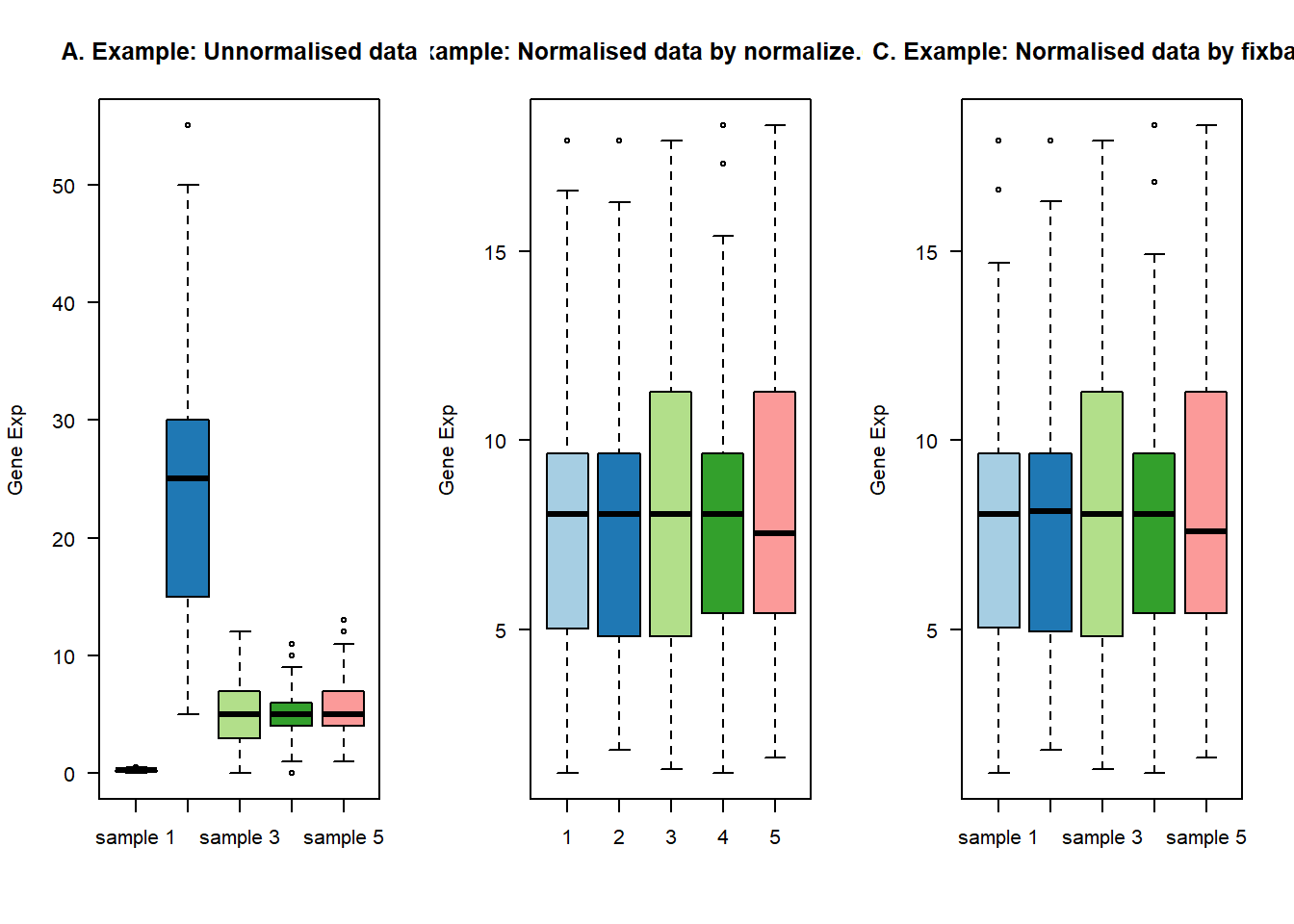
看起来效果还不错,然后雄赳赳,气昂昂的去实际操作,实践是检验标准的唯一真理!发现我就发现其没有任何效果!!!,不论是用我自己的R包,还是经典的preprocessCore包。都是没有效果的!!!
dat_df = get_df(dat)#change into data.frame
#dat_df = column_to_rownames(dat,vars = id)dat_fix2 = norm_fix(dat_df,expcv = 0.3, exphigh = 0.8, explow = 0.4)## The stable housekeep gene number is 6507# dat_fix2 = norm_fix(dat_df,expcv = 1, exphigh = 1, explow = 0)
# 设置expcv = 1, exphigh = 1, explow = 0等同于原始的preprocessCore包!
dat_pca_fix = pca_data_tidy(dat_fix2)## ... Notice: the input data is a data frame not a tibble
## ... Wonderful: the input data vaule in every column all be numberic value
## ... Successed! the pca data save in the objectp2 <- pca(dat_pca_fix,rename = "replace",
str_sample = "_fpkm",
str_group = "-\\d$",
display_sample = T,
add_ploy = T) +
ggtitle("Normalised dat")
print(p1+p2+plot_annotation(tag_levels = 'A'))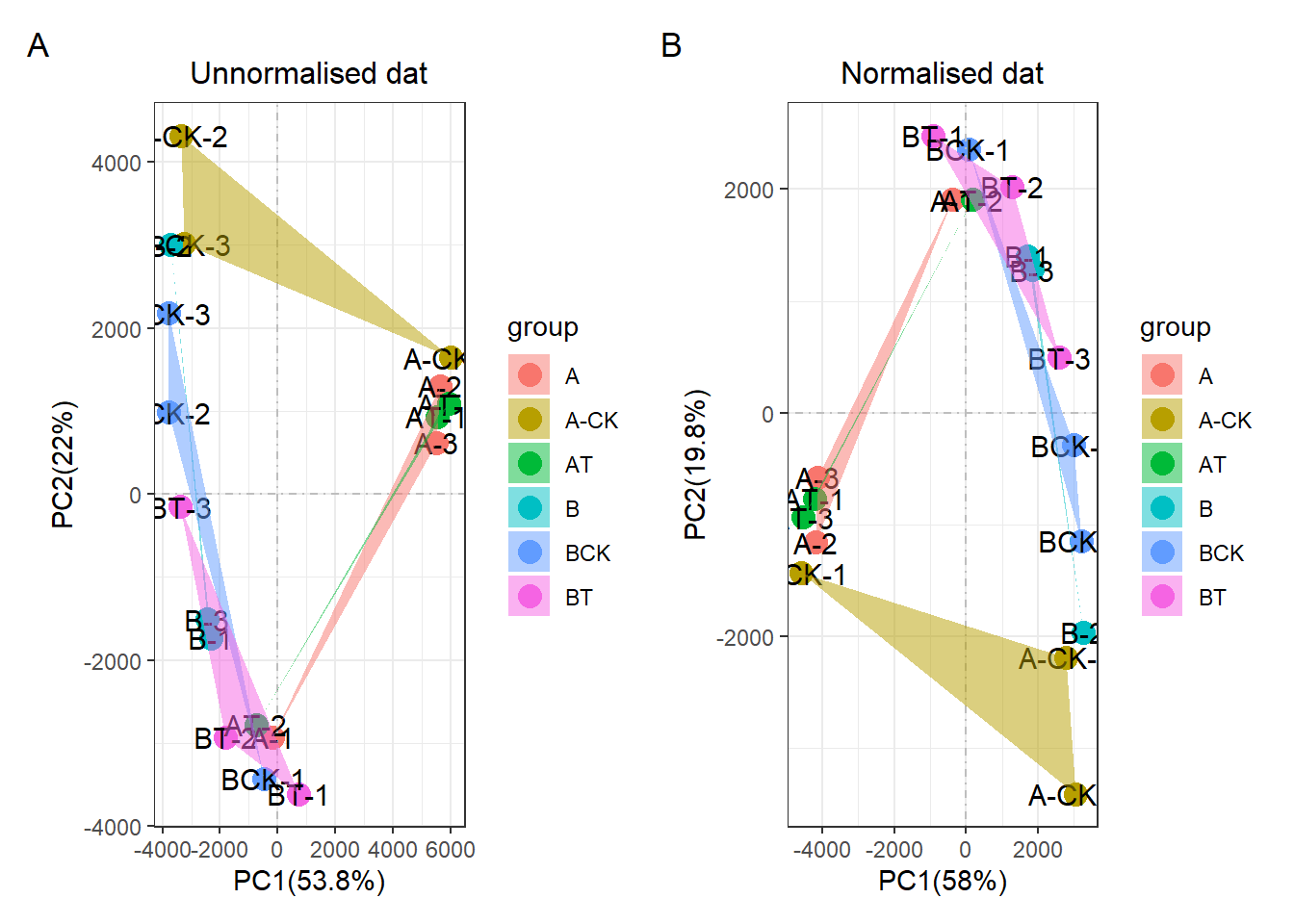
后续的思考
所以这个R包是失败的!我心好凉,我后续在这个R包里面加上edgeR的矫正方法,以及sva的矫正方法。这两个R包肯定是有用的!!!
preprocessCore失败,我觉得是没用考虑生物问题。但我本来以为我利用管家基因的来计算矫正因子是正确的思路。但为什么结果也还是失败呢。
今天在写公众号的时候,我突然想到,我对管家基因的定义是有问题的。
因为我选择变异系数低的,以及表达量中间的基因,并不真正是管家基因!!甚至说,因为变异系数低,所以不论是离群样本,还是正常样本,我所挑选的管家基因,表达量是不会变化的,所以矫正因子在实际项目中,大概率永远都是1左右徘徊。
我应该换思路:
- 增加指定内参基因的功能,例如指定GAPDH,TBP,UBC,ACTB等基因
- 优化内参基因的选择方法,根据批次分组来分别选择,再取交集,计算矫正因子
等我改好了,倒是再看看效果。
加油,干饭人!