一般转录组数据因为其通量高。一下检测几万的基因表达量,面对如此丰富的表达量信息。就为根据表达量的情况来筛选那些内参基因提供了一个很好的研究基础。
背景
在一些实验设计,老师比较关注的是在不同品系间的内参基因。类比于18s和actin,内参基因首先的定义就是不受内外因素的影响,表达量趋向恒定的一类基因。虽然这些基因一般都是一些非常重要以及**“基础”**的管家基因。虽然一般做qPCR等实验,用18s、GAPDH、actin这些内参基因就足够了。但是一般转录组数据因为其通量高。一下检测几万的基因表达量,面对如此丰富的表达量信息。就为根据表达量的情况来筛选那些内参基因提供了一个很好的研究基础。
言归正传,如何根据基因的表达量来进行**内参基因的筛选呢。
目前,我的常用方法是计算Unigene的离散系数CV。简单讲就是CV=标准差/平均数,数值越小,这个基因在不同样本间的表达量的变化越小。

这个定义可以看
变异系数是衡量资料中各观测值变异程度的另一个统计量。当进行两个或多个资料变异程度的比较时,如果度量单位与平均数相同,可以直接利用标准差来比较。如果单位和(或)平均数不同时,比较其变异程度就不能采用标准差,而需采用标准差与平均数的比值(相对值)来比较。标准差与平均数的比值称为变异系数,记为CV
当然,这个CV来计算,并得到内参基因,是否有文献支持,说实话,因为我没有这方面的研究背景,所以并不清楚,只是从统计学上讲,CV是非常不错的筛选标准。
如果各位老师,有更好的统计参数来衡量和筛选,非常欢迎与我说明,不胜感激!但是目前看, CV值是个很不错的标准, 甚至有人为此写了一个R包. 有兴趣的老师可以去看一下这边文章.

用R来计算CV
今天的主要内容是如何利用R来计算CV,以前,我会选择用excel来进行计算CV
其实excel计算起来并不复杂,用函数STDEV()和AVERAGE(),就可以了,只需要计算两个值的比值就可以。
但是为什么用R来计算,因为R首先是专业的统计软件,(并不是绘图软件!!!),并且用R会显得有逼格嘛,哈哈
其实主要是写一个脚本,以后遇到这样的分析,就可以自动跑出来了啦,这是最重要的
为什么大家会认为R是绘图软件,我觉得这锅要Haddly来背(ggplot2的作者)
开始用R之前
因为,我的R主要是靠自学和看书,我看的第一本是Haddly的《ggplot2:图形绘画与艺术》,第二本是Haddly的《R for data science》,目前第三本估计还是Haddly的《advance R》。
兄弟,发现问题了没。所以我的R命令很多都依赖Haddly的R包。但其实Haddly只是众多R开发者之一,虽然无疑Haddly是改变R的人,但是R是一个社区,很多其他人R包也是很优秀的。所以我的命令有的时候就很不协调和冗余,这是因为为了兼顾Haddly的包特有的格式。比如tibble和R自带的data.frame之间的行名之间的冲突。今天我才稍微知道了colunm_to_rownames命令如何协调两者。
所以各位看官,看我的命令一定要知道为什么这样,以及换成简单命令。或者你也学学Haddly的R包吧
命令行
在开始之前,推荐一个IDE和文本编辑器。
sublime文本编辑器
在此之前,之前有个老师向我推荐了sublime这个文本编辑器,这个编辑器真心牛逼,针对R的脚本,实现了非常美观的代码高亮。非常nice
另外,在安装一些插件后,就可以变成R的IDE,这个非常完美了。我即嫌弃Rgui的简陋界面,又嫌弃Rstudio的繁琐界面。
终于在安装sublime后,既有Rgui的简洁界面,又有Rstudio的代码补全。
我好喜欢这个sublime,哈哈哈
用R计算离散系数
注释看代码行
# R 3.5.2 win 10
require(tidyverse)
require(matrixStats)#这个包有rowSds函数,计算一行的sd标准差
path = "C:/Users/woney/Desktop" # 这是我的工作目录桌面
inname = "all.sample.annot.xls"
setwd(path)
dat=read_tsv(inname, col_names = T)
nms <- str_subset(colnames(dat),pattern = "pm|km")
dim(dat)
dat_m <- dat %>% filter(rowSums(.[nms]) != 0) %>%
mutate(mean = rowMeans(.[nms]),sd = rowSds(as.matrix(.[nms]))) %>%
mutate(CV = sd/mean)
dat_cv <- dat_m
write_tsv(dat_cv,"unigene_cv.xls",col_names = T)
dat_quan <- quantile(dat_cv[["CV"]],prob = 0.01)#寻找排名前1%的最小CV值基因
dat_cv_fit <- dat_cv %>% arrange(CV) %>% filter(mean >= 100, CV <= dat_quan)
write_tsv(dat_cv_fit,"unigene_cv_filter.xls",col_names = T)
到此,我们就得到了一个完整结果的文件,一个我们筛选后的表格,但是感觉还是缺一个图形。
绘制图形
对于这个目标结果的可视化,我没什么好的经验。所以暂且用散点图来绘制,但是感觉不太匹配,要是有好的建议,可以分享一下,一起学习和进步。
#绘制图形
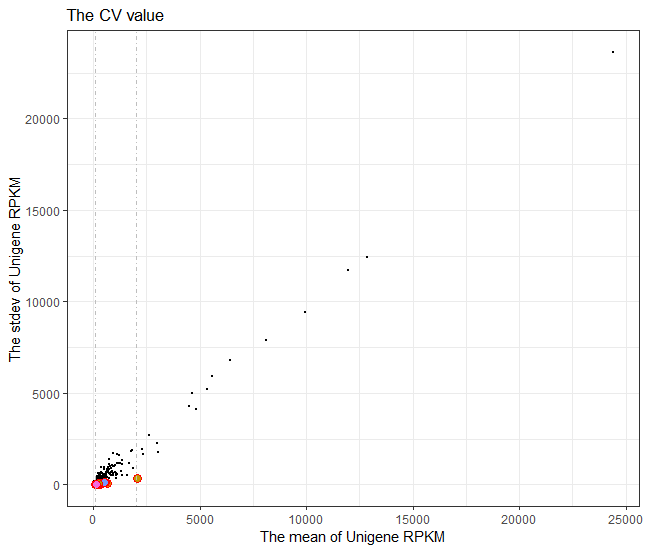
p1 <- ggplot(dat =dat_cv_fit, mapping = aes(x = mean, y = sd))+
geom_point(dat = dat_cv,aes(x = mean, y = sd),fill = "skyblue",size = 0.5)+
geom_point(dat = dat_cv_fit,aes(fill = id),shape = 21,color ="red",size = 3)+
labs(x = "The mean of Unigene RPKM",y = "The stdev of Unigene RPKM" ,title = "The CV value")+
geom_vline(xintercept = c(100,2000),linetype = 4,color = "grey")
#进行一点调整
p2 <- p1 + theme_bw()+ theme(legend.position = "none")
检验结果
毕竟是自己的脚本,R实现的优势是速度快,但是只要其中一个命令出错,就可能导致结果的差异。对于自己不熟悉的东西,尤其是计算类的,还是小心一点比较好。
所以我在excel上,还是检查了一下,我的结果。还是一致的。可靠!
| id | mean | sd | sd | CV |
|---|---|---|---|---|
| Unigene0047702 | 105.8204 | 8.716806 | 8.716806 | 0.082374 |
| Unigene0071345 | 110.7748 | 11.24985 | 11.24985 | 0.101556 |
| Unigene0024038 | 235.0149 | 26.4493 | 26.4493 | 0.112543 |
| Unigene0054875 | 224.0919 | 26.61498 | 26.61498 | 0.118768 |
| Unigene0003494 | 143.8802 | 18.38708 | 18.38708 | 0.127794 |
| Unigene0001501 | 136.5825 | 17.94474 | 17.94474 | 0.131384 |
| Unigene0061046 | 132.6634 | 17.51517 | 17.51517 | 0.132027 |
| Unigene0051467 | 224.9567 | 29.70607 | 29.70607 | 0.132052 |
| Unigene0002648 | 635.3467 | 84.06525 | 84.06525 | 0.132314 |
| Unigene0018474 | 315.5729 | 43.3001 | 43.3001 | 0.137211 |
| Unigene0064436 | 179.2159 | 24.72725 | 24.72725 | 0.137975 |
| Unigene0058797 | 126.3847 | 17.8765 | 17.8765 | 0.141445 |
| Unigene0053692 | 139.5825 | 20.52212 | 20.52212 | 0.147025 |
| …………….. |
后言
其实,用R做离散系数的分析,就到此完结撒花了。
其实,这个过程是挺曲折的,但是至少在这个过程中,增加了对R的理解。