了解一下甲基化
重亚硫酸氢盐测序(BS-Seq)或全基因组亚硫酸氢盐测序(WGBS)就是我们常说的甲基化测序,目前是用于检测基因组*DNA中的胞嘧啶甲基化的最成熟方案。在该方法中,用重亚硫酸氢钠处理基因组DNA,经过测序,在基因组水平上提供胞嘧啶甲基化的单碱基分辨率。在亚硫酸氢盐处理时,未甲基化的C脱氨基成U*,U在测序后转化为T。同时,甲基化的C拒绝脱氨作用,并被测序为C。然后可以通过比较处理和未处理的序列来确定甲基化胞嘧啶的位置。DNA的亚硫酸氢盐处理将未甲基化的C转化为T,导致序列复杂性降低,这对SBS合成系为原理的illumina二代测序来说,是非常不利的。所以需要非常准确的深度测序才可以减轻这种复杂性的损失。
优点:
- 整个基因组中的
CpG和非CpG甲基化以单碱基分辨率覆盖 - 覆盖密集,密度较小和重复区域的
5mC
缺点:
- 亚硫酸氢盐将未甲基化的胞嘧啶转化为胸[^1]苷,降低序列复杂性,这使得难以产生比对
- 在亚硫酸氢盐转化时,错过胞嘧啶转化为胸苷的
NPs - 亚硫酸氢盐转化率不区分
5mC和5hmC
我的非覆
Methods Link:
- BS-Seq: Feil R., Charlton J., Bird A. P., Walter J. and Reik W. (1994) Methylation analysis on individual chromosomes: improved protocol for bisulphite genomic sequencing. Nucleic Acids Res 22: 695-696
- Bisulfite-seq: Berman B. P., Weisenberger D. J., Aman J. F., Hinoue T., Ramjan Z., et al. (2012) Regions of focal
DNAhypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 44: 40-46- WGBS: Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., et al. (2009) Human
DNAmethylomes at base resolution show widespread epigenomic differences. Nature 462: 315-322
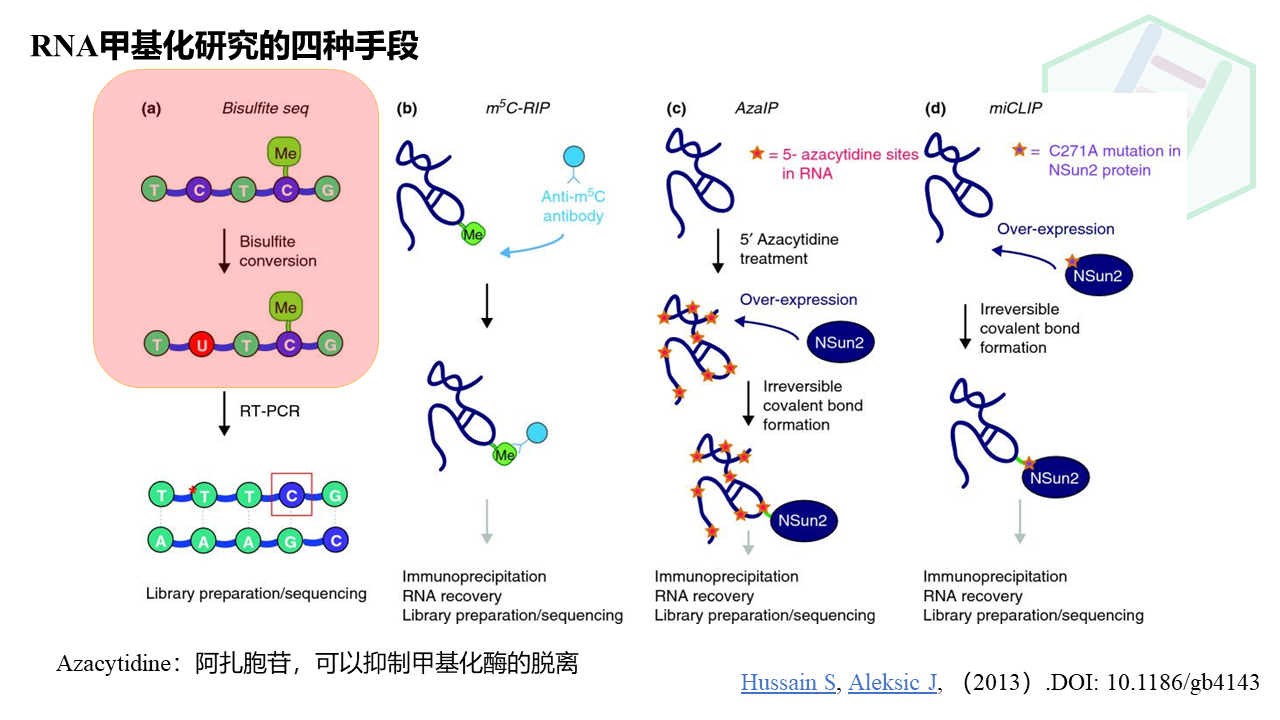
图中展现的是RNA的四种甲基化研究手段,但是红色框内的同样适用于我们今天重点讲的*DNA*的胞嘧啶甲基化。
WGBS全基因组甲基化测序
得到* DNA* 样品后,首先对样品进行质量检测。样品质量检测合格后,进行 BS 文库构建,具体步骤如下:
- 基因组 *
DNA*超声打断成100-300bp的片段 DNA片段末端修复、3’端加 A 碱基,连接测序接头;- 采用 ZYMO EZ
DNAMethylation-Gold kit 进行Bisulfite处理; - 脱盐处理后切胶回收,并进行文库片段大小选择,PCR 扩增后再次进行文库片段大小选择;文库构建完成后,对文库进行质量检测。质检合格的文库将用于上机测序。
将测序结果与参考基因组比对,比对上唯一位置的序列用于后续标准信息分析及个性化分析。得到下机数据后,首先进行数据过滤,去掉低质量数据,得到可用数据。完成数据过滤后,需检测可用数据量是否符合合同要求。检测合格后,将可用数据与参考基因组进行比对,得到比对结果。在确认比对质量合格后,使用唯一比对数据得到全基因组* C* 碱基甲基化信息,进行信息分析处理,得到标准信息分析结果和个性化分析结果。
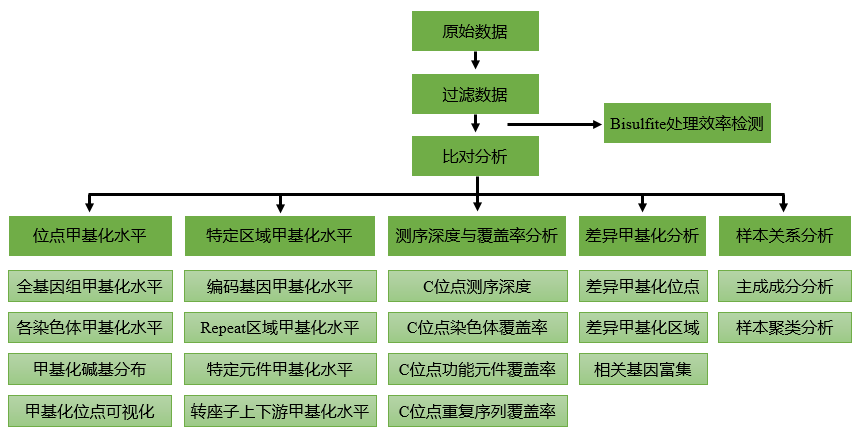
以上实验和分析步骤主要来自基迪奥公司的结题报告,官网:http://www.genedenovo.com/
质控标准
相比于其他流程,甲基化测序的非常特殊的一点是,需要加入lambda DNA(lambda噬菌体DNA)进行Bisulfite(重亚硫酸盐)处理效率检测。这是因为理论上,lambda DNA上的C均为非甲基化的C,在Bisulfite处理后理论上全部C将被转化成U(即测序中的T),但处理转化失败的位点,则会依然保留C不变。
所以统计C位点被转换成T的比率就可以得知重亚硫酸盐的处理效率,一般要大于99%。
分析内容
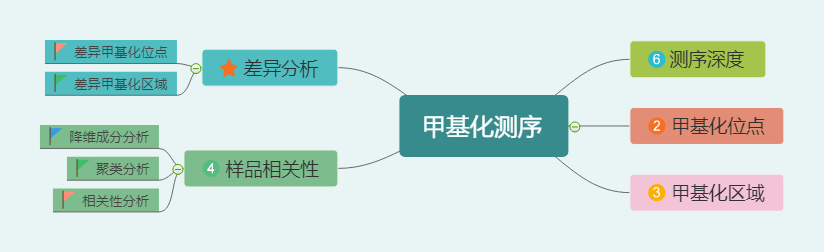
甲基化的分析内容很多,尤其又分为三种甲基化位点(CG, CHH, CHG),三个作用区域(UP, DOWN, BODY),还有两种分析思路(甲基化区域和甲基化位点),以及各种容易混淆的概念。比如:
你能分清甲基化位点这四种概念嘛?
- 甲基化位点的数量:发生甲基化的 C 位点的数目
- 甲基化位点的比例:甲基化 C 位点数目 / 总 C 位点
- 位点甲基化值(水平):发生甲基化的 C 位点,支持发生甲基化的 reads 数 / 测到这个位点的 reads 数目
- 所有位点的平均甲基化值:在基因组水平上,所有发生甲基化的 c 位点的位点甲基化值(水平)的平均值。
仔细一一说,实在太久了,简单总结下:
我最近再看一篇《Genome Biology》的关于草莓发育的甲基化文章,《Genome Biology》自然就不用多说了,组学期刊中的数得着。而且我最近发现很多甲基化的相关的文章都是《Genome Biology》,不知道是不是我搜索的关系。
文章解读

《草莓果实成熟期间RdDM的下调》
数据探索
草莓果实的DNA甲基化组织
为了研究草莓的甲基化水平,作者取了全果实(包括叶和果实,果蒂)进行了全基因组亚硫酸氢盐测序,得到了DNA甲基化的单碱基分辨率图。从未成熟阶段到成熟阶段(Fa1-Fa3)的果实用两个生物学重复序列测序(图 1a)。

由于栽培的八倍体草莓(F. ananassa)和野生二倍体草莓(F. vesca)之间的高度共线性,以及F. ananassa八倍体栽培种缺乏组装良好的基因组,我们使用F. vesca的基因组作为参考。在我们的分析。F. vesca的基因组约为240 MB(2 n?= 14)。对于每个测序文库,产生至少100M配对末端读数(大约就15G的数据的量,60x的覆盖度),基因组覆盖> 80%,大约10%的reads被定位到未甲基化的叶绿体基因组,并且重亚硫酸氢盐的转化率> 99.6%。对每个甲基化组进行测序,染色体平均覆盖率 > 10倍。本次测序的结果展示,测序覆盖度和深度和前人研究的拟南芥和番茄甲基化的实验相差无几。
未成熟草莓果实的平均DNA甲基化率约为7.5%,低于番茄果实(22%),mCG,mCHG和mCHH的平均甲基化水平分别为40%,11%和2%。在植物中,转座因子(TE)和重复序列通常是高度甲基化的。为了研究草莓中DNA甲基化的基因组分布,作者使用RepeatScout从头注释了F. vesca基因组中的TEs和重复序列。草莓基因组中的TE和重复含量高于拟南芥,但低于水稻和番茄,与较大基因组往往具有较高重复序列比例的猜想一致。
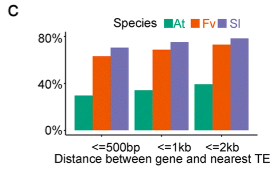
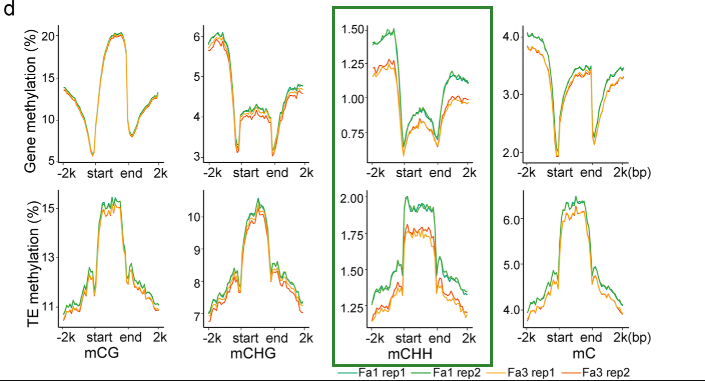
叶片的平均mC水平约为8%,略高于未成熟果实(7.5%)。叶子和果实在基因和TE周围表现出相似的DNA甲基化模式,但一般而言,叶子相对于果实具有更高的DNA甲基化水平。在基因密集的区域,甲基化水平低,而在转座子密集的区域,甲基化水平高。如下图:

根据前人的研究,基因在中型大小基因组的物种中,如番茄,基因与转座子具有更短的距离,距离越近,代表越有可能受到转座子的调控。而基因组小的物种,比如拟南芥,就会有更长的距离。这个假设是符合生物学逻辑的,但是在八倍体草莓中,这个基因与转座子的距离,却比番茄还短。作者认为可能是因为基因组的组装不好的问题(最强解释)。
草莓成熟期间DNA甲基化的丧失
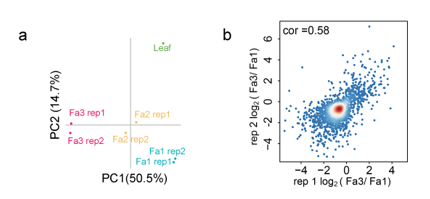
在番茄成熟期间DNA甲基化显着降低,为了研究草莓果实成熟过程中的DNA甲基化动力学,我们在三个不同阶段(Fa1-Fa3)比较草莓果实的DNA甲基化组,每个阶段有两个生物学重复。主成分分析(PCA)显示每个阶段的两个生物重复之间的一致性。我们发现,成熟过程中提示在DNA甲基化降低。这种减少发生在两个生物学重复中。
同时作者也证明了在编码基因的转录起始位点(TSS)上游2kb的一直到转录终点(TES)后2kb的区域内,成熟过程中Fa3相比于Fa1,DNA甲基化水平降低。
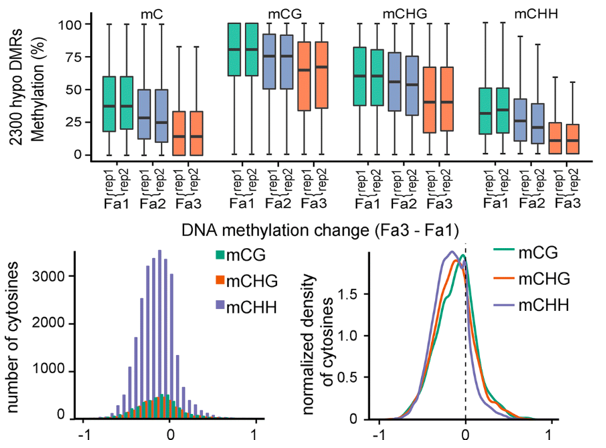
作者比较了Fa1和Fa3时期的果实甲基化水平,差异分析显示,一共有2766个DMR区域。其中2300个是下调的,466个DMR是发生上调的。对每一个生物学重复的差异变化的值,进行相关性分析,结果显示相关性强(智者见智吧)。
作者为了探究甲基化水平的下调是否与甲基化类型相关,就分别看了三种甲基化类型之间的变化。结果表明,草莓在成熟过程中三个甲基化类型都会发生DNA甲基化水平的降低,这点和番茄类似。
验证甲基化数据
基于以上结果,作者提出了在果实的发育过程中,甲基化水平的调控是起着非常重要的作用,一方面为了探究甲基化水平的调控机制,一方面又为了验证甲基化的数据,为后续的数据挖掘提供一个实验结果的支持。所以用DNA甲基化抑制剂5-azacytidine,处理草莓幼果。与空白处理相比,5-azacytidine处理的果实表现出早熟的表型。为了验证5-azacytidine处理,是否通过抑制果实中的DNA甲基化水平影响果实成熟,我们对空白组和5-azacytidine处理组,进行methylation-sensitive qPCR。
qPCR验证了两个基因,证明5-azacytidine处理的草莓果实,发生了显著的DNA甲基化水平降低。这些结果表明DNA低甲基化对草莓果实成熟很重要。

这个methylation-sensitive
qPCR很有意思,值得去研究一下实验方法,这个验证的基因数目,你懂的,不过表型从文献的图片上看是很明显的。
甲基化位点的基因组分布
在Fa1 -vs- Fa3比较组中,作者评估了DMR在基因组上的分布情况。随着草莓果实的成熟,作者发现在转录起始位点(TSS)上游区域的甲基化水平发生显著的降低。
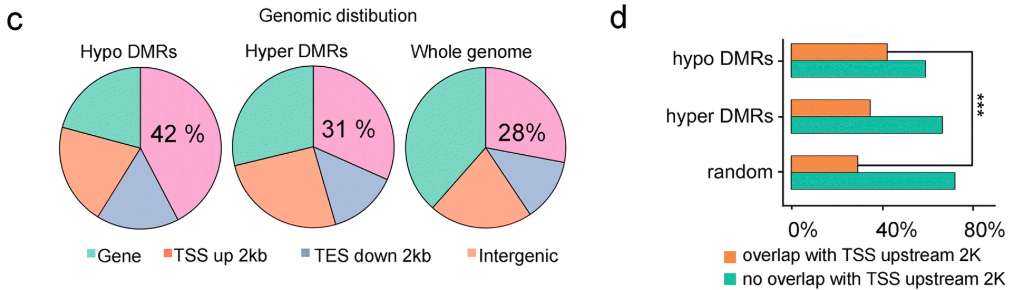
同时,作者也发现差异降低的DMR主要集中在基因的5'-和3'-区域中。相反,差异升高的DMR的分布没有表现出这样的模式。这些结果表明,成熟过程中的DNA低甲基化可能广泛参与到基因的调控过程中。
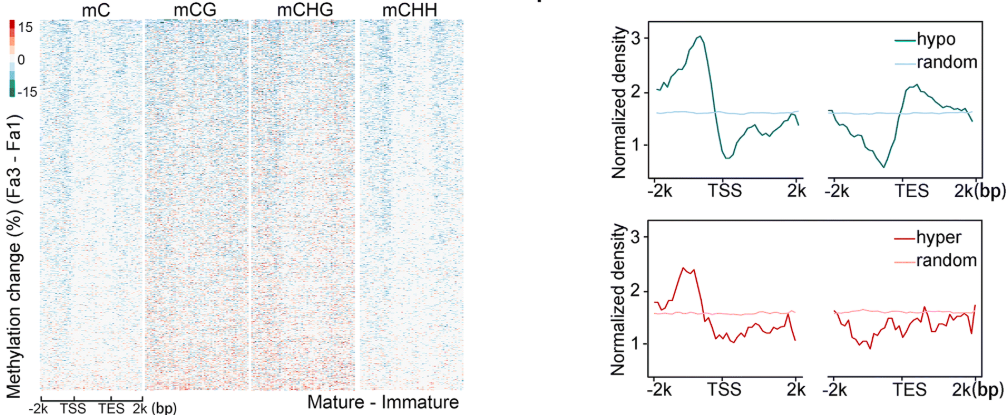
到此作者发现了随着果实成熟,甲基化水平是整体上下降的。那么是是什么机制导致 这样的生物学现象呢,所以就做了这方面的探究,去更加深入的阐述这一生物学现象。
成熟过程中RdDM途径基因的表达量降低
在番茄中,成熟过程中DNA甲基化水平降低是由于DNA去甲基化酶SLDML2的表达量升高。在拟南芥中,主要是通过ROSR家族蛋白完成对5mC的清除。此外,IDM蛋白复合物可以调节ROS1的活性,促进去甲基化过程。
在草莓中,发现这些基因在草莓中的同源基因,都没有在成熟过程中发生显著上调(作者没说,其实是下调的0.o)。这些结果表明草莓成熟期间的DNA低甲基化与DNA去甲基化途径基因的表达增加无关。
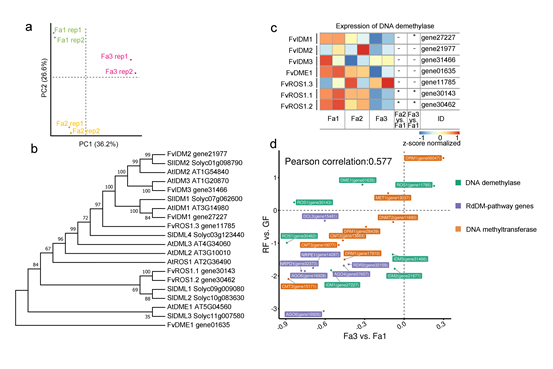
去甲基化研究的进度不好,那就从甲基化入手,DNA去甲基化和DNA甲基化相互拮抗,动态调节植物DNA的甲基化水平。作者假设降低的DNA甲基化活性可能有助于草莓果实成熟期间的DNA低甲基化。为了验证这一假设,我们检查了DNA甲基化途径中涉及的基因的表达。
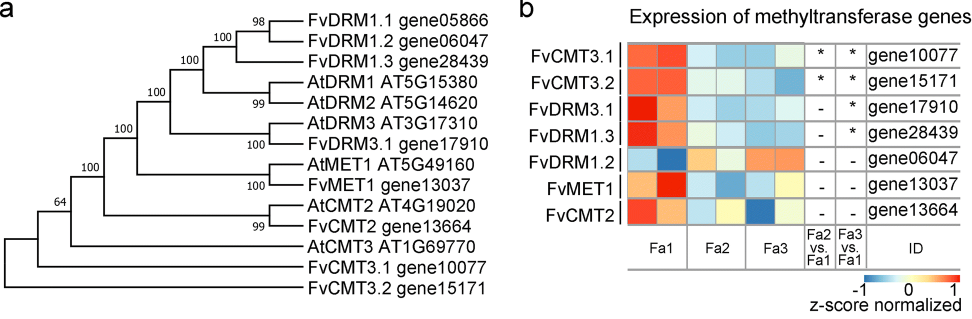
在拟南芥中,mCG,mCHG和mCHH分别由甲基化转移酶1(MET1),CMT3和CMT2和重链甲基化酶(DRM)维持。
同时,所有三种甲基化情况都可以通过由RNA介导的,从而影响DNA发生甲基化。 称为RdDM途径。
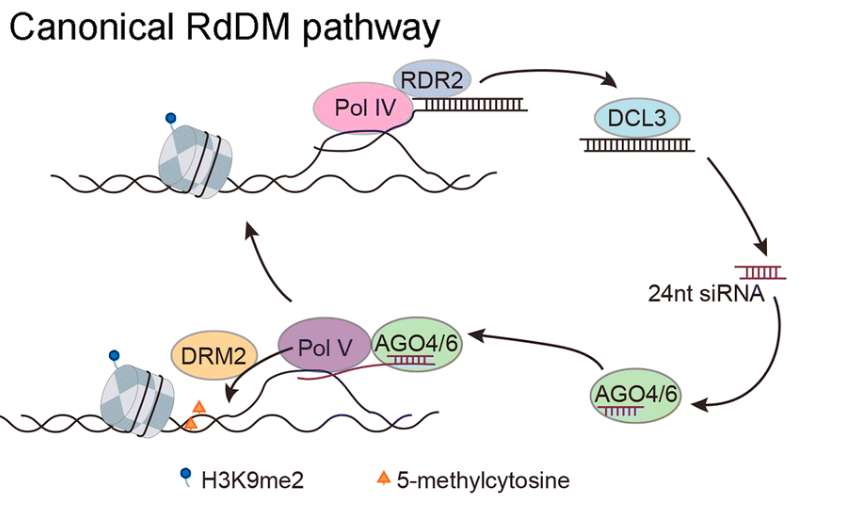
通过RNA指导的DNA甲基化(RdDM)途径,主要有两个主要步骤:第一步是诱导产生siRNA。第二步,就是由siRNA引导发生DNA甲基化。RNA聚合酶IV转录组产生siRNA的前体,再通过DCL3(DICER-like家族蛋白)剪切前体形成成熟体的siRNA。在第二步骤中,siRNA可以结合ago家族蛋白去调控DRM甲基化蛋白的转录活性,从而完成对DNA的甲基化修饰。DNA甲基化状态可以通过DNA甲基转移酶和去甲基化酶动态调节。
作者在草莓基因组中鉴定了8个DNA甲基转移酶基因,ok,随着成熟发育,基因的表达量符合预期。这些数据表明减少的DNA甲基化活性有助于成熟诱导的DNA低甲基化。
siRNA在成熟发育中诱导DMR的差异下调
上文刚刚提到了siRNA街道的RdDM甲基化途径,作者紧接着就做了sRNA测序。sRNA的长度分布和首位U碱基都得到了很好得验证(测序中A),说明测序质量合格。
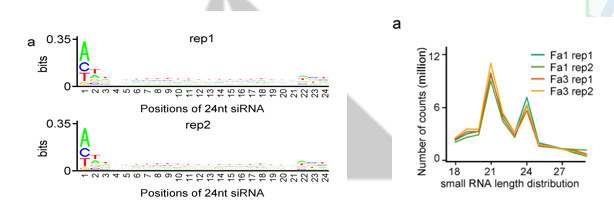
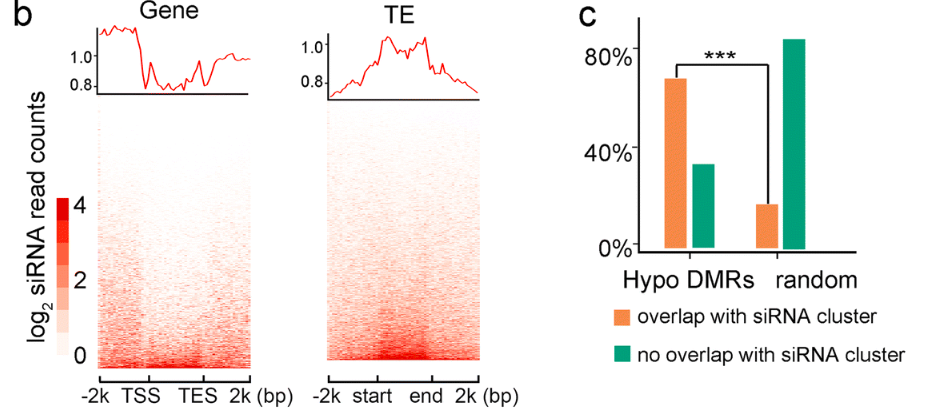
和甲基化分析部分一样,作者对sRNA得位置分布,以及和随机分布情况进行差异显著性分析。其实这篇文章我唯一不清楚得就是如何算出来这样一个随机数值(测序的数据已经有自己的分布,难道是随机从基因组中截取24nt的片段,抽取10w次不等来模拟随机的嘛),总之,作者发现sRNA主要分布在转录本的上下游区域,代表着sRNA肯定在调控基因表达中起着重要的作用。同时作者验证了与随机事件相比,成熟期间发生差异下调的DMR区域中有67%以上区域与siRNA簇有overlap。
相比之下,只有17%的随机基因组区域与siRNA簇重叠,这表明成熟诱导的低DMRs与siRNAs显着相关(p ?<0.01)
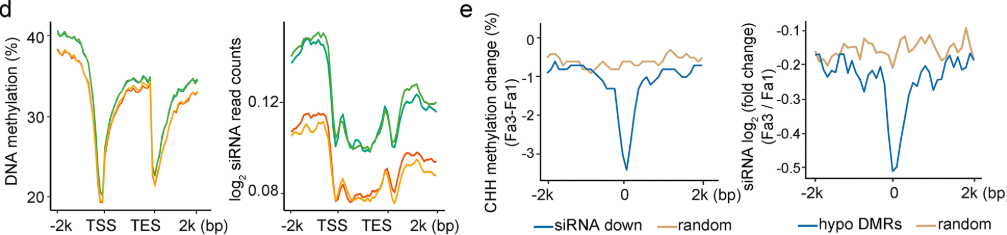
为了进一步研究siRNA介导的DNA甲基化与成熟发育中的DNA甲基化水平降低之间的关系,作者监测了成熟的低甲基化的DMRs中siRNA富集的情况。发现,siRNA的表达量水平从FA1到FA3降低。
同时,发现siRNA的变化情况和甲基化水平的变化情况一致,这个图还是有点抽象,不过作者说什么就是什么啦。
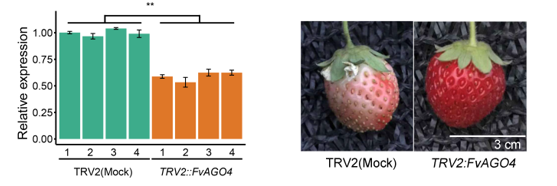
为了测试RdDM途径对草莓成熟的重要性,作者使用TRV介导的基因沉默来下调幼草莓果实中的FvAGO4。我们在TRV2::FvAGO4果实与对照仅TRV2果实相比,观察到早期成熟表型,这一表型与RdDM在草莓成熟中活性降低导致低甲基化水平的猜测一致。这些分析表明,RdDM途径基因的表达量下调,导致siRNA积累减少,最终导致RdDM活性降低,从而促进DNA低甲基化和草莓成熟。
这才是生物学故事啊
DNA甲基化变化与果实中基因表达的改变有关
为了研究成熟诱导的DNA甲基化变化与基因表达之间的关系,作者做了Fa1和Fa3的果实转录组。一共有2316个差异表达的基因(adj_Pvalue <?0.01),其中包括899个上调DEGS,和1417下调的DEGS。
接下来,检查了上调和下调DEG的DNA甲基化水平。我们发现,DEGs中的的大部分基因都是经历DNA甲基化水平从FA1到FA3降低(不知道你们能不能看出来,反正一眼扫过去,确实是蓝色的多一点)
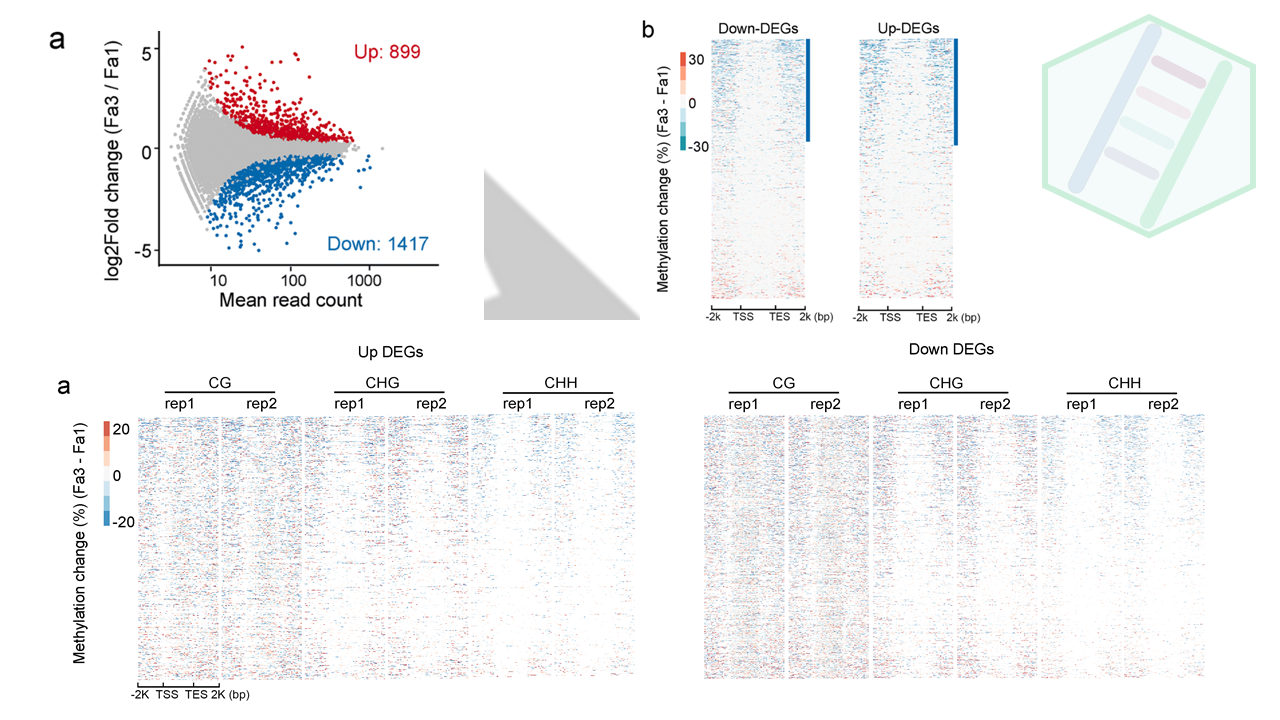
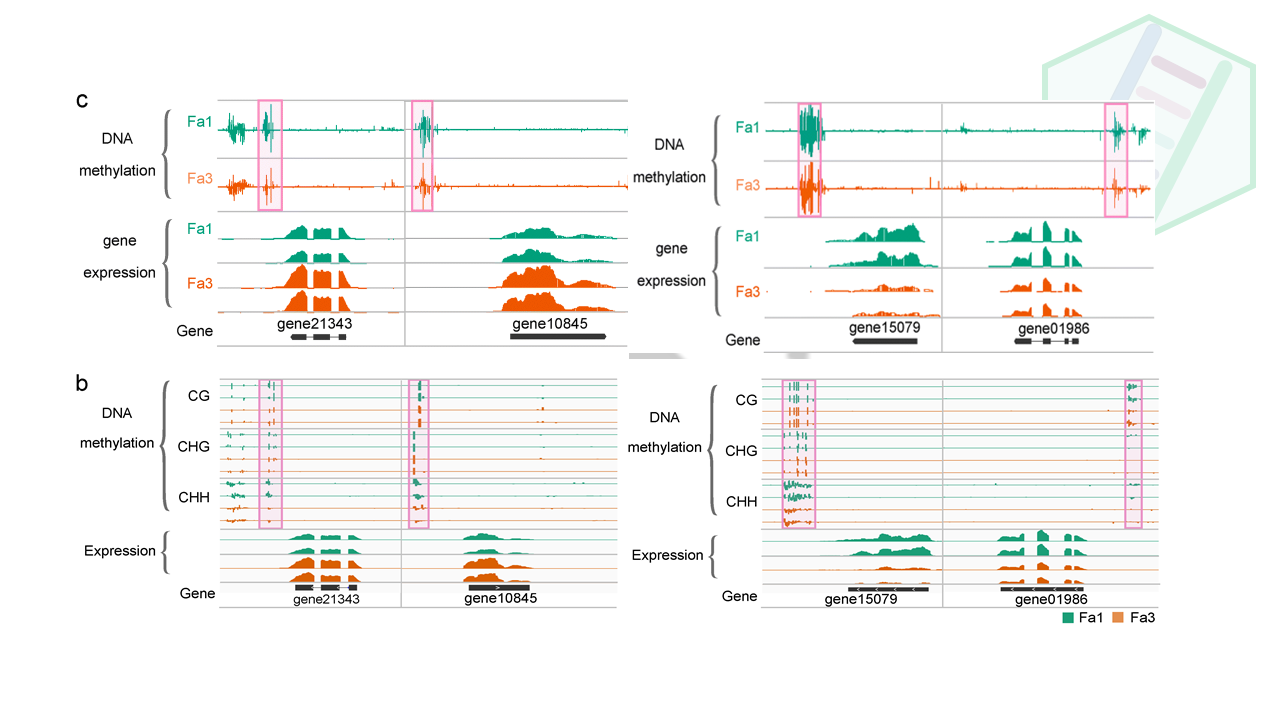
这里是整体上描述了差异变化基因DEGs的差异甲基化情况,作者之前不是做了两个me-qPCR的基因嘛,正好拿来讨论一番,一来凑个内容,二来局部到点,彰显自己细致的研究。通过IGB图来展现基因的表达量与甲基化、sRNA之间的关系。
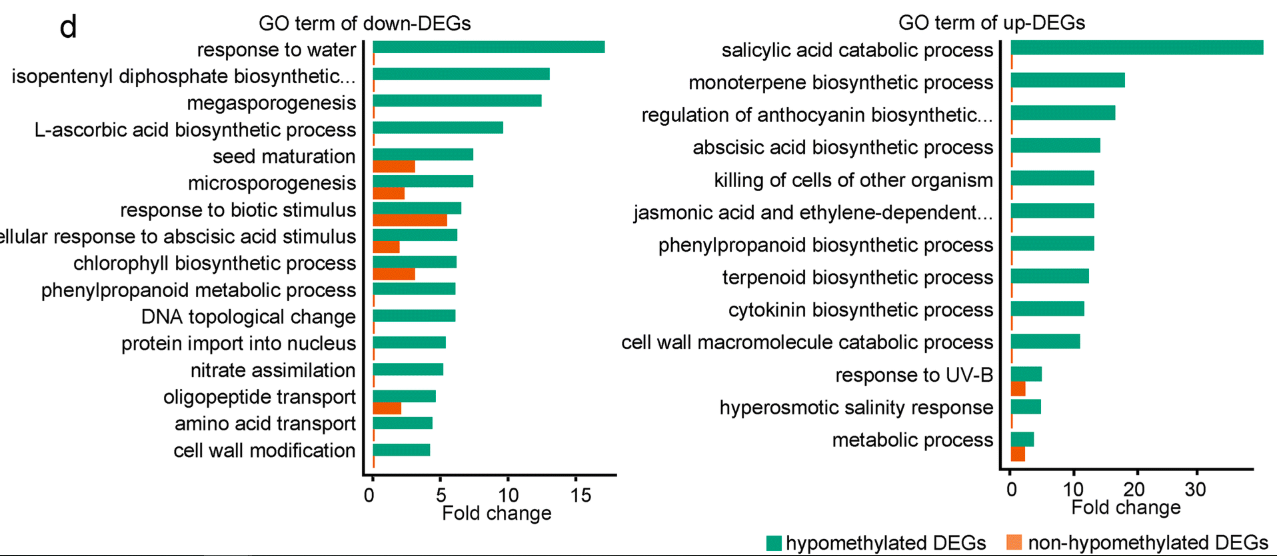
既然知道差异基因,不做个富集分析,简直就是对不起组学文章这个头衔啊。做完富集分析之后,就要讨论生物学功能,这个才是考验老师的写作能力的重点。要不然富集分析谁都会做,但是经常遇到老师问,我怎么挑选,你能帮我挑选吗。自己的课题自己一定要把握啊,这么能让别人来挑。
Fruit ripening is a complex developmental process that involves numerous physiological, biochemical, and structural alterations. To understand how
DNAmethylation-associated gene regulation contributes to the ripening process, we performed Gene Ontology (GO) analysis for hypomethylated up- and downregulated DEGs. Up- and downregulated DEGs that are not hypomethylated served as controls (Fig. 6d). This analysis revealed that genes involved in the “regulation of anthocyanin biosynthetic process” are enriched in upregulated DEGs (Fig. 6d and Additional file 7: Table S6), suggesting thatDNAmethylation may regulate fruit coloration during ripening. A previous study found that gene21343 (expansin-A8-like) is required for biosynthesis of the pigment anthocyanin during strawberry ripening [25], although the underlying molecular mechanism was unclear. Our results showed that gene21343 displayed decreasedDNAmethylation during ripening, and its expression was upregulated accordingly (Fig. 6b, Additional file 2: Figure S6b). Cytokinin is important for early fruit development, and a recent study suggested that cytokinin accumulates during the ripening of kiwi, grape, and strawberry fruits [26]. Consistent with these findings, our analysis found that genes involved in the “cytokinin biosynthetic process” are enriched in the upregulated DEGs (Fig. 6d and Additional file 7: Table S6). The endogenous content of ABA is known to increase substantially during strawberry ripening, and the application of an ABA synthesis inhibitor interrupts strawberry ripening [8]. Consistent with these observations, we found that genes involved in the “abscisic acid biosynthetic process” are enriched in the upregulated DEGs (Fig. 6d and Additional file 7: Table S6). Several other ripening-related GO terms were also included in the upregulated DEGs, such as genes involved in the biosynthesis of flavor volatiles (Fig. 6d and Additional file 7: Table S6).On the other hand, many genes need to be downregulated during ripening. For example, genes involved in photosynthesis are active and required in the early stages of fruit development, but are repressed during ripening to allow conversion of chloroplasts into chromoplasts, which are important for fruit coloration. The hypomethylated downregulated DEGs were enriched for “chlorophyll biosynthetic process,” suggesting that
DNAhypomethylation may contribute to the decline in photosynthesis during ripening. We also found several GO terms related to cell wall biosynthesis and metabolism in hypomethylated downregulated DEGs. During early fruit development, genes involved in cell wall biosynthesis and modification are active to sustain fast cell division and cell enlargement, but these genes are not needed later during the ripening stage. Interestingly, the ripening-related GO terms were only enriched in hypomethylated DEGs, but not enriched in up- or down-DEGs without hypomethylation (Fig. 6d and Additional file 7: Table S6). Together, these analyses suggest thatDNAmethylation-mediated gene regulation controls many biological processes important for ripening.
材料与方法
实验材料
物种:草莓(Fragaria×ananassa Duch.cv.Hongjia)
**参考基因组:**双倍体Fragaria vesca v1.1https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Fvesca
产地:浙江省海宁市浙江省农业科学院科研创新基地
取样时期:三个发育阶段的果实
- Fa1,绿色阶段,开花后约20天(DAB);
- Fa2,中间红阶段,30DAB;
- Fa3,全红阶段,35DAB
在收获当天转移到实验室后,选择具有均匀尺寸且无可见缺陷的果实。每个发育阶段使用8-12个果实混浑混合。两个生物学重复送去测序。
对于DNA甲基化抑制剂处理,使用“Si gongzhu”草莓果实。在处理中,将溶解在含有0.01%Triton X-100的ddH 2 O中的20mM 5-阿杂胞苷(Sigma)直接喷洒在果实上。处理于6月8日,13日和18日进行。样品于6月23日拍摄。
甲基化PCR
根据试剂盒的说明,用McrBC消化基因组DNA(100ng)12小时。没有GTP的消化用作阴性对照。在65℃下酶灭活20分钟后,将10%的消化DNA用于定量实时PCR的每个PCR反应。
全基因组亚硫酸氢盐测序和数据分析
文件构建
使用DNeasy Plant Maxi Kit(Qiagen)从叶和果实中提取基因组DNA。
使用Illumina HiSeq2500对样品进行测序。使用用于Illumina的NEBNext Ultra II DNA文库制备试剂盒和Epitect Plus DNA Bisulfite Kit(Qiagen)制备全基因组亚硫酸氢盐测序文库。
简言之,将1μg基因组DNA在Covaris M220上超声处理成200~500bp片段。
按照NEBNext Ultra II DNA文库制备试剂盒的制造商说明,对片段化的DNA进行末端修复,A尾和接头连接。
然后使用Epitect Plus DNA Bisulfite Kit(Qiagen)在热循环仪上用亚硫酸氢钠处理接头连接的产物,具有以下程序:
- 95°C 5分钟,
- 60°C 25分钟,
- 95°C 5分钟,
- 60°C 85分钟,
- 95°C 5分钟,
- 60°C 175分钟
- 95°C 5分钟,60°C 180 min,三个循环
- 冷却至20°C
然后使用Epitect Plus DNA Bisulfite Kit(Qiagen)清洁BS处理的DNA,并使用KAPA HiFi Hotstart ReadyMix进行PCR扩增6个循环。最后使用来自Vazyme的磁珠清除扩增的文库。
使用HiSeq PE Cluster Kit v4(Illumina)和HiSeq SBS Kit v4(250个循环)(Illumina)按照制造商的说明在HiSeq2500(Illumina)上以双端125bp模式对文库进行测序。
数据分析:
对于数据分析,首先使用Trimmomatic修剪末端测序读数,去除Illumina衔接子和低质量碱基(质量评分<20)。然后使用bsmap-2.87 将clean reads比对到Fragaria vesca的基因组上。采用默认的参数设置。
用methratio.py(bsmap-2.87中包含的脚本)提取甲基化比率。
仅考虑在所有文库中具有至少4个深度的胞嘧啶。窗口大小为200bp,步长为50bp的滑动窗口方法用于识别DMR。对每个窗口内的甲基化与未甲基化胞嘧啶进行Fisher精确检验。使用Benjamini-Hochberg调整后估计错误发现率(FDR)值。FDR <0.05的窗口被定义为进一步分析的候选DMR。
然后通过在所有三种情况下合并候选物并缩小至第一种和最后一种差异甲基化胞嘧啶(DMC)来调节DMR。用组合标准过滤最终的DMR:DMC数> 3,平均甲基化差> 0.15。为了将生物复制品纳入分析,我们计算了鲁棒指数作为每个DMR的公式,以测量重复之间的重复性。 $$ \mathrm{Robust}\kern0.5em \mathrm{index}=\frac{\left|{\log}_2{FC}_1-{\log}_2{FC}_2\right|}{\left|{\log}_2{FC}_1+{\log}_2{FC}_2\right|} $$ FC 1和FC 2分别代表重复1和重复2中的差异甲基化倍数变化。具有较低鲁棒指数的DMR在差异甲基化方面更可靠。将单个重复中具有低深度的DMR的稳健指数指定为NA。我们通过稳健指数对DMR进行排名, 并分别重新分析前500和至少500可信的低DMR。
RNA测序和数据分析
用来自水果的TRIzol试剂(Ambion)提取总RNA。对于逆转录,使用qScript cDNA SuperMix试剂盒(Quanta),使用1μgRNA和oligo dT引物在20-μL反应中合成cDNA。对于RNA-seq,使用Illumina HiSeq2500在中国科学院上海植物胁迫生物学中心的基因组学核心设施中构建和测序文库。
对于数据分析,使用STAR 将配对末端读数与Fragaria vesca基因组。使用默认参数比对。FeatureCounts 用于计算每个基因的映射片段(count值,不过这个软件第一次见)。DESeq 用于检测差异表达的基因。为了在Fragaria vesca中分配假定的基因功能,通过Phytozome数据库寻找同源基因。
小RNA测序和数据分析
用来自水果的TRIzol试剂(Ambion)提取总RNA。然后在变性聚丙烯酰胺凝胶上分离总RNA,切出<100-nt级分并纯化用于标准小RNA文库制备。对于小RNA测序,使用Illumina HiSeq2500在中国科学院上海植物胁迫生物学中心的基因组学核心设施构建和测序文库。
用Trimmomatic 修剪测序读数以去除含接头和低质量碱基(质量评分<20)。长度<18bp且长度> 30bp的读数被丢弃。24-nt读数被定位到F. vesca基因组并用Shortstack定义为siRNA簇[ 36 ]。通过总清洁读数对映射的读数进行标准化以进行进一步分析。
Shortstack :这个鉴定sRNA簇的软件值得留意
系统发育分析
使用Phytozome注释的DNA甲基转移酶,DNA去甲基化酶和参与RdDM途径的基因的直系同源物构建了与MEGA相邻的系统发育树。
常规操作
病毒诱导的基因沉默(VIGS)
Liu等人描述了用于基因沉默的TRV载体pTRV1(pYL192)和pTRV2(pYL156)实验方案如Birch-Smith等人所述。将pTRV2载体与FvAGO4的 PCR片段连接,然后转化到A.tumefaciens GV3101菌株中。分别用pTRV1,pTRV2和pTRV2衍生物pTRV2- FvAGO4转化土壤杆菌培养物(在25mg / L利福平和50mg / L卡那霉素过夜培养物中生长)。将土壤杆菌培养物渗入草莓的果梗中。
没构建TRV质粒,不清楚,不过原理都是一样的