新的Python脚本出炉了
这个功能就是为了实现从大量的输出文件中提取某一列的内容到新的汇总表格中,应用场景一般是分批跑组学样本,导致每个样本都有一个输出文件。这个时候需要提取某一列信息,生产总表。
常见的比如宏基因组的Metaphlan输出,转录组的Stringtie输出等等吧,总之我是遇见了几次,所以才把这需求写成一个脚本。方便下次遇到的时候使用。
输入文件
举个例子:假设我现在手上有一个耐药基因输出的表格如下: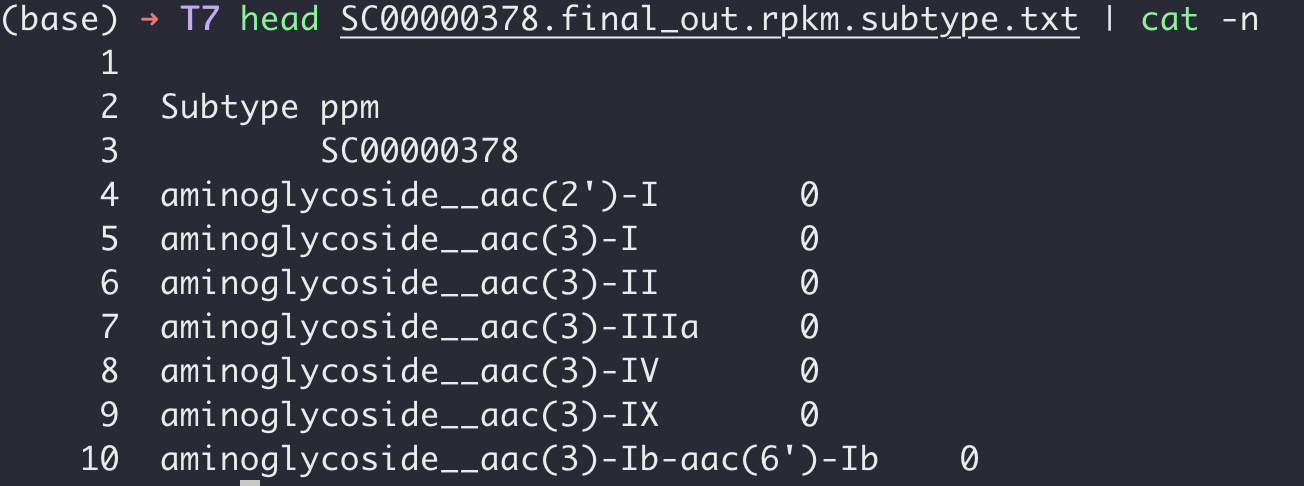
可以看到这个文件的格式是:
- 第一行是空白,第二行是无用信息,第三行才是这个样本SC00000378的样本名称,也就是我们常认为可以做表头的信息。
- 只有两列,第一列是耐药基因,第二列是耐药基因表达量
那么上图是一个样本的信息,假设我们有三百个样本的信息,又该如何去写代码?
在我们不知道每个样本的耐药基因顺序是否会发生变化的时候,我想没人敢直接合并在一起。所以最安全的方法还是要在合并的同时,还要根据耐药基因的名称进行匹配合并。才是最准确的做法。
解决思路
如果用R来做
我能想到的是,用list.files()枚举出所有文件,所有构建循环:readr::read_csv -> dplyr::full_jion,这样如此不断的A->D->C->….这样去递推到最后一个文件。
有没有更好的R的方法来做这个事情呢,希望评论区有大神能有更好的思路建议
如果用Python
因为一般组学的流程分析都是大型集群上,在远端终端上输入命令行。这种情况下,如果不是遇到很复杂的需求,我更宁愿去学shell,而不是进行python或者R的界面进行操作。因为感觉不够流程吧。
所以我将python脚本写成命令行运行的方式,参数的含义在帮助文章里写了出来,如下图:
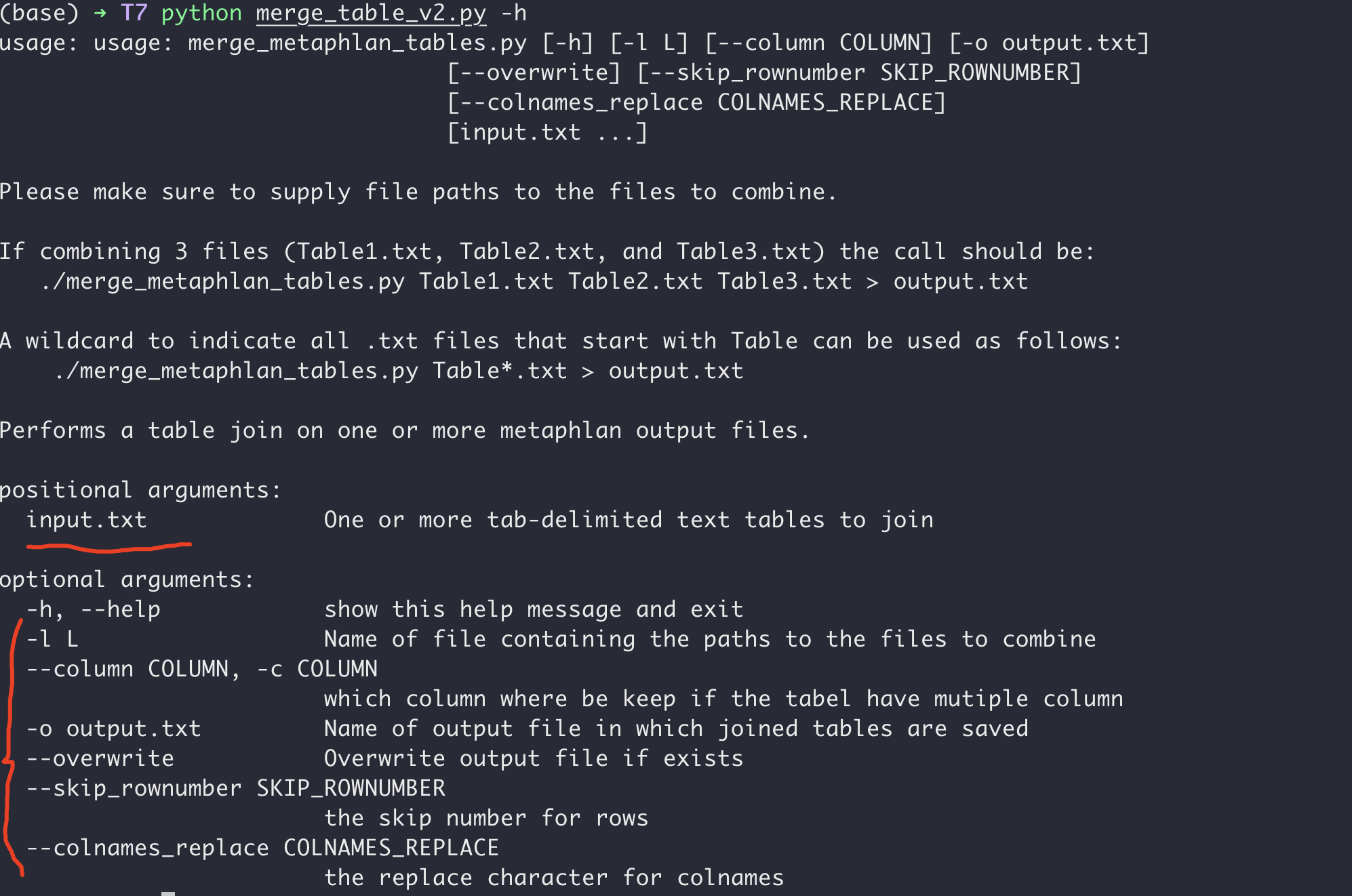
简而言之:就是-c指定那一列进行提取合并,--overwrite决定是否覆盖同名文件,-o 是输出的新文件(同时也支持标准std.out),--skip_rownumber是决定前多少行不读取。--colnames_replace比较特殊,是因为这个脚本会将文件名当成该文件提取列的新列名,所以一般需要去掉一些文件后缀。所以才有这个。举个例子SC00000378.final_out.rpkm.subtype.txt合并后,SC00000378.final_out.rpkm.subtype.txt也会作为列名存在新表格中,但是有了--colnames_replace .final_out.rpkm.subtype.txt,那就删掉匹配字符,只留下SC00000378。
运行
假设我们现在三个样本的耐药基因输出表格(如第一张图一样的格式)
ls *.final_out.rpkm.subtype.txt
#SC00000378.final_out.rpkm.subtype.txt SC00000553.final_out.rpkm.subtype.txt SC00000858.final_out.rpkm.subtype.txt
合并这三个文件,参数需要有:
- 前两行没用:–skip_rownumber 2
- 第一列是行名,第二列需要提取(pandas data.frame 中0就是除行名外的第一列) -c 0
- 去除名称后缀,–colnames_replace .final_out.rpkm.subtype.txt
- 当然还有输入和输出了
python merge_table_v2.py *.final_out.rpkm.subtype.txt -o test.tsv -c 0 --skip_rownumber 2 --colnames_replace .final_out.rpkm.subtype.txt --overwrite
当运行成功后,新文件就如下图所示了:
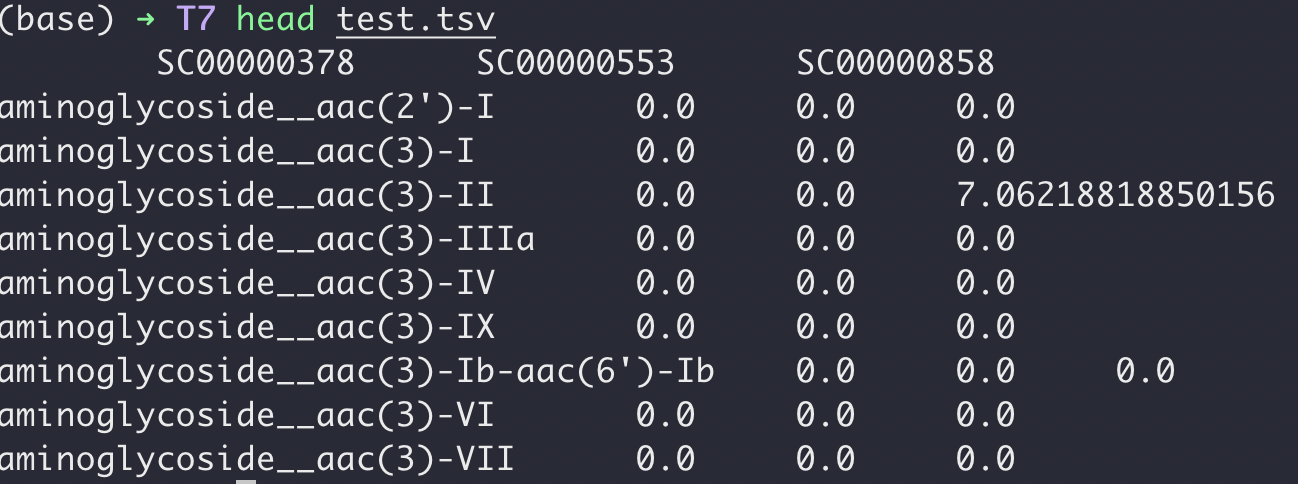
完美!
写好的脚本我放在Github:wangjiaxuan666/WeChat-pubword/merge_table_v2.py上,微信可点击原文链接,需要的可以下载,请随意。