做整合分析的时候,看到broad研究所大佬👨写的软件太好:,看了很久源码,学习wdl,自我感觉小有所成,重构下之前用shell搭建的甲基化流程😸。
BSseq-WDl流程介绍📖
背景 🌁
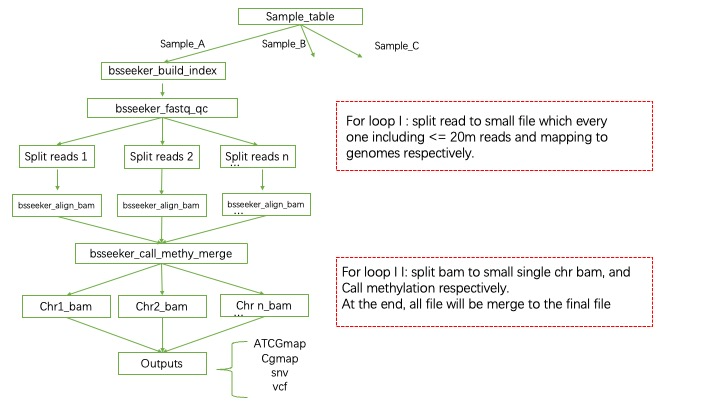
BSseq-WDl 是我根据 BS-seeker2 + CGmapTools 为基础搭建的甲基化分析主要流程。说是主要,其实就是输出到甲基化结果文件(甲基化位点和甲基化率)。
该流程的优点🏆
说这个流程的优点,其实就是说wdl的优点,相比于之前用shell写的。我觉得wdl最大优点就是:
- 彻底不用管文件的归档和存放,文件的输出和输入实现流程化控制
- 任务的并行运行,以及多个任务的多次调用比较方便
- 搭建虽然辛苦,但是维护起来比较简单
该流程的缺点📕
但是说完优点,也要说缺点,真的也是有点难以忍受
- 没有积极活跃的社区,用户少,意味着问题很大概率要自己debug,让人崩溃
- workflow,call,task 三者都要一个input,再加上一个output。一直让人来回输入那几个变量(因为要变量承上启下,还要声明),繁琐且容易报错,让人没有感觉到一点代码的简洁之美。
- 文件放在特定的目录下,层级太多,很难找到,必须搭配输出的json文件
- if 语句中竟然还要评估否条件下的表达式,离谱!
其他的缺点暂时想不到了,如果一句话概括就是:
过程很痛苦,结果很美好,可能是流程控制语言的通病吧
流程空间流程📦
用wdl编写流程,需要cromwell.jar才能运行,输入文件有两个:
- 🌴参数输入文件:*.json
- 🌵样本名称和fq路径表格文件:*.tsv
输出文件需要添加 -m 参数,详情请见运行部分,输出的json文件: 包括了:
- 过滤后的read
- mapping后的用于call methylation的bam文件
- 甲基化结果文件
*.wig,*.ATCGmap,*.CGmap - call snp的结果文件
*.snv,*.vcf
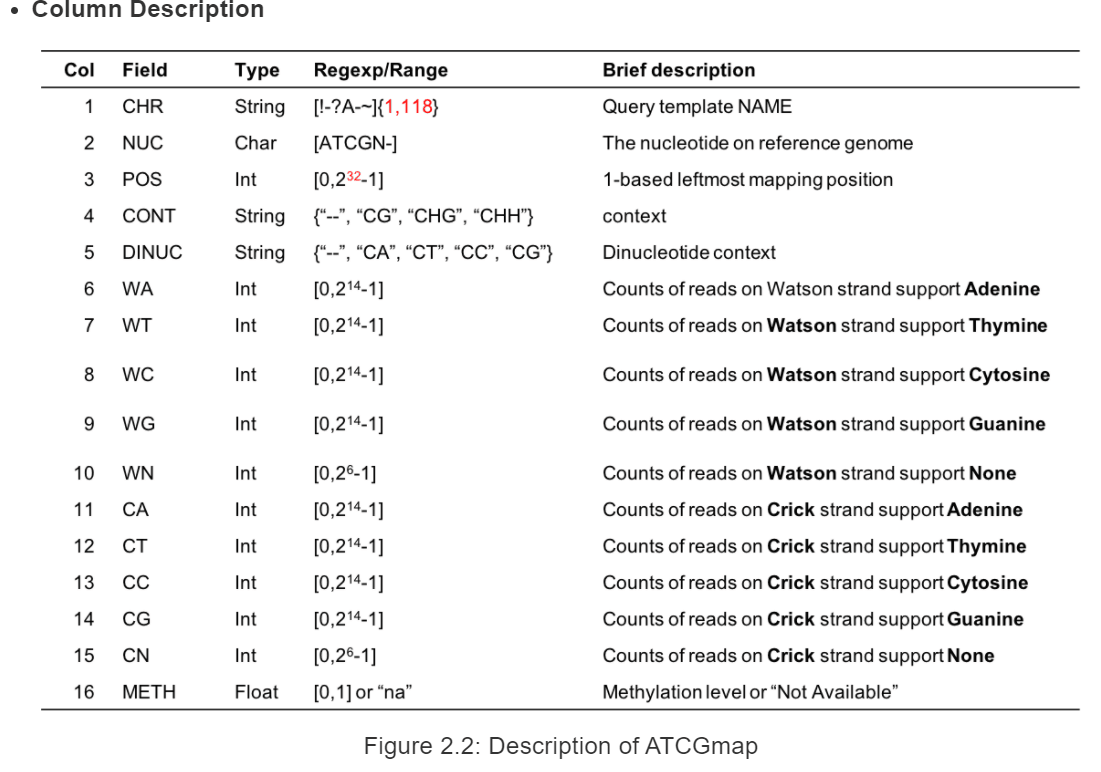
其中*.ATCGmap,*.CGmap是主要输出结果,前者代表是多种类型的甲基化位点结果(CHH,CHG,CG),后者只有CG类型的甲基化位点信息。结果是表格形式的,分别存储有位点,绝对甲基化reads数和相对的甲基化率等数值。
流程采用并行的方法,大幅提高了运行速度和准确性
环境要求🍄
本流程运行需要以下环境:
- 需要python 2.7(pysam)
- 需要java 1.8.0 的环境
- 环境中要有samtools,fastp,CGmapTools环境变量
- 需要下载BSseeker2
关于BSseeker和CGmapTools的安装示例问题,可以参考后文
流程运行🐚
软件下载
下载Github仓库,解压,在联网环境下,运行download_cromwell.sh脚本,下载cromwell-58.jar☕,如果已经有cromwell☕其他版本的可以不用下载,可以自行尝试是否版本兼容。
bash download_cromwell.sh
配制文件
输入文件有两个,一个控制参数的json文件,一个是记录样本名称和fq序列文件路径的tsv表格.
tsv 表格
制表符分割的文件,有三列,分别是样本名称,read1的路径,read2的路径。
Example:
sample_A ./test/sample_A.test.1k.R1.fq.gz ./test/sample_A.test.1k.R2.fq.gz
sample_B ./test/sample_B.test.1k.R1.fq.gz ./test/sample_B.test.1k.R2.fq.gz
暂不考虑单端测序和生物学重复,前者是用不到,后者是还没做差异
json文件
需要一些参数才能运行:
{
"bsseq.genome_index" : "error_path_just_for_test/test.fa_bowtie2/",
"bsseq.python_file" : "wangjiaxuan/biosoft/miniconda3/envs/py27/bin/python",
"bsseq.bsseeker_util_dir" : "wangjiaxuan/biosoft/BSseeker",
"bsseq.refer_fa" : "./test/test.fa",
"bsseq.alig_software" : "bowtie2",
"bsseq.fastq_table" : "./test/input_sample_fq.tsv",
"bsseq.step02_fq_qc" : "true",
"bsseq.step03_bsseeker_align" : "true"
}
其中genome_index是基因组索引文件,这个是要BSseeker构建的。如果之前没有构建过,这个参数可以不写或者给了错误的地址。**流程都会自动根据参数refer_fa来进行基因组索引构建,只是需要的时间和内存都会比较大。
另外参数refer_fa是必须参数,除此以外,必须的参数还有python2.7路径的python_file,BSseeker的脚本目录bsseeker_util_dir,输入上文提到的tsv表格的fastq_table,以及比对软件alig_software。
运行
方法一🐍
运行流程, 我写了一个python的外包,运行./bsseq-run -i bsseq.input.json就可以开始分析,参数-i控制输入的json文件,-c是指定的cromwell.jar的路径,默认是在WDL文件下的。
# 参数
# usage: bsseq-run [-h] --input INPUT [--cromwell CROMWELL] [--version]
# BS-seq analysis workflow base on bsseeker and CGmaptools
# optional arguments:
# -h, --help show this help message and exit
# --input INPUT, -i INPUT
# the input json
# --cromwell CROMWELL, -c CROMWELL
# Optional path to cromwell jar file
# --version, -v show version tag: 0.1
方法二 ☕
当然外包的python又需要python3的环境,而运行BSseeker是python2的环境,我也没无奈,因为我不会python2,所以嫌麻烦也可以直接运行命令行:
java -jar WDL/WDL/cromwell-58.jar run WDL/bsseq.wdl -i bsseq.input.json -m bsseq.output.json
# -m 参数是输出文件,可以不写,但是不写后续分析很麻烦
结果解读🍵
结果可能有点疑惑,但这也是wdl的特性,输出文件都会在cromwell-executions文件里,里面根据task来进行分文件,又进一步细分为input和execution。但是看bsseq.output.json就不会疑惑了。
有用的信息,我都output了
Example:
"outputs": {
"bsseq.bsseeker_call_methylation_bam_bai": ["sort_rmSX.bam.bai", "sort_rmSX.bam.bai"],
"bsseq.bsseeker_call_methylation_bam": ["sort_rmSX.bam"],
"bsseq.sample": ["sample_A", "sample_B"],
"bsseq.fastp_filter_fq1": ["sample_B.filter.R1.fq.gz"],
"bsseq.bsseeker_methy_CGmap": ["merge.CGmap"],
"bsseq.bsseeker_index": "test.fa_bowtie2",
"bsseq.bsseeker_methy_wig": ["merge.wig"],
"bsseq.cgmaptools_methy_snv": ["bayes.snv"],
"bsseq.fastp_filter_fq2": ["sample_B.filter.R2.fq.gz"],
"bsseq.cgmaptools_methy_vcf": ["bayes.vcf"],
"bsseq.bsseeker_methy_ATCGmap": ["merge.ATCGmap"]
}
# 信息匿去部分信息,本身是按照样本输出的
甲基化结果文件展示:🍸
我们拿ATCGmap文件举例,结果是文本格式,包含甲基化位点和甲基化水平 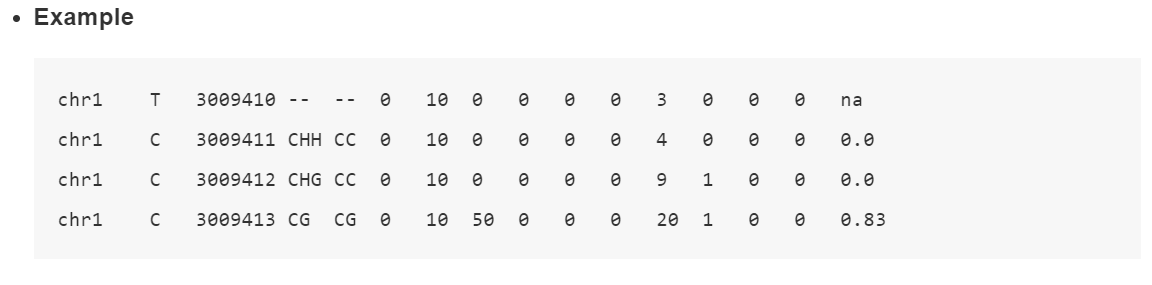
具体每一列的详细信息如下:
安装 ⛵
安装BSseeker2
按照BSseeker的网址上的介绍安装和使用 简单点话就是
git clone https://github.com/BSSeeker/BSseeker2
cd BSseeker2
就可以看到其中的py脚本,但是亲测python 3.8不能用,只能用conda创建一个python2.7的环境,同时需要环境中有bowtie, bowtie2, soap三个软件中任意一个都可以。python环境也要有pysam模块。
安装CGmapTools
git clone https://github.com/guoweilong/cgmaptools
bash install.sh
# add cgmaptools to your system PATH if necessary
# open ~/.bashrc
vi ~/.bashrc
# add the following lines to the end of ~/.bashrc
export PATH=/path/to/cgmaptools/:$PATH
# then, souce your ~/.bashrc
source ~/.bashrc
还需要在python2.7的环境中安装pysam和scipy
conda activate py27
pip install pysam
pip install scipy
资料来源⚓
资料主要来源黄湘仪的教程——18.10.04亚硫酸盐测序数据基本分析流程代码及软件,当然也感谢BSseeker2 和CGmapTools开发者农大的郭伟龙博士(中国农业大学)和朱平博士(天津血研所/北京大学)等人合作开发了DNA甲基化数据分析工具包:CGmapTools,包含40个独立的命令行工具,提供了全面的DNA甲基化数据分析和可视化功能。目前该工作已经在生物信息学著名期刊Bioinformatics上以original paper形式发表。
Weilong Guo , Ping Zhu , et al. (2017), CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data.
再次感谢各位大佬创建这么好用的软件