一篇Nat Rev Genet 的文章《Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics》,关于单细胞和空间组数据分析
最近经同事介绍里一篇Nat Rev Genet 的文章《Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics》,是关于空间组和单细胞如何互相关联分析的。虽说我感觉其中思路并没有非常大的创新,但是也是一篇比较好的归纳总结。

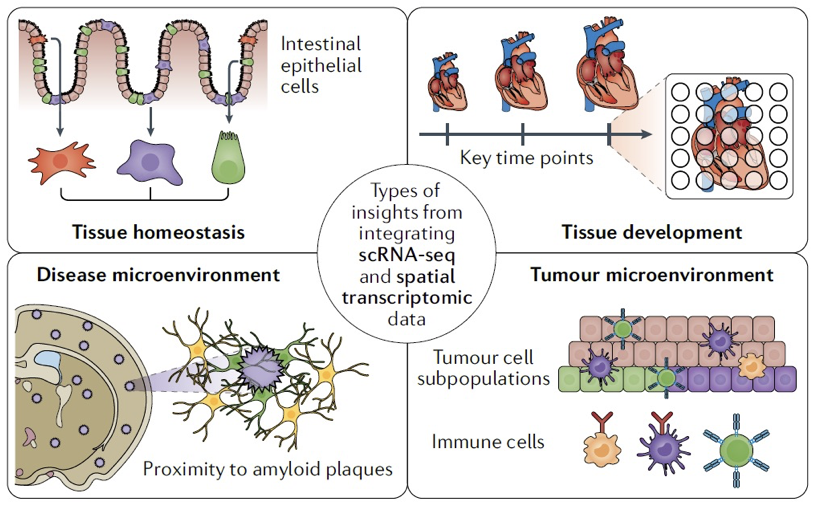
空间组可以解决的生物学问题
作者提出一个比较新颖的名词Tissue homeostasis,姑且先称之为组织稳态吧,作者将组织稳态分别分为:
- 1️⃣ Tissue development
- 2️⃣ Disease microenvironment
- 3️⃣ Tumour microenvironment
Tissue development是指研究在组织发育的关键阶段,空间转录组如何在组织中发生变化。Disease microenvironment是指阐明患病和受伤组织生态位中的空间转录组,着眼于接近相关的生物学特征,例如,接近阿尔茨海默病患者脑组织中的淀粉样蛋白斑块。Tumour microenvironment是指研究肿瘤的空间结构及其与环境中其他细胞亚型的界面和交流。
感觉每个方面都是非常重要的生物问题,而也都非常适合空间组数据来进行解释和分析,所以空间组的分析目前应该还算是蓝海吧。
文中还列举了一些文章的例子
正常组织发育
空间转录组学的一个主要目标是增加对不同细胞类型在稳态组织稳态中的作用的理解。例如,沿着血管周围桡骨轴的每个肝小叶单元都含有不同的氧气,营养物质和激素梯度,再结合scRNA-seq揭示了一类新型中间小叶细胞的作用,FISH验证定位,结束(Natrue 2017)。
肿瘤微环境
TSK定位到人鳞状细胞癌肿瘤边界处,验证其功能(Cell 2020)。在胰腺导管腺癌中,揭示炎症成纤维细胞在癌症应激反应中起重要作用(NBT 2020)。数据还可以提供对临床预后的见解。例如,一项研究观察到,黑色素瘤转移横截面内过渡区域的异质性越大,患者生存期越差(Cancer res 2018)
其他患病和受伤组织的微环境
除了肿瘤相邻的微环境,空间组此外还可以检测其他疾病的外部环境。将空间组数据与免疫染色的成年小鼠大脑横截面对齐,确定了淀粉样蛋白斑块密度较高的区域中细胞共有的基因网络。用单细胞分辨率图谱,并捕获它们在不同疾病阶段的空间动态。阐明与淀粉样蛋白斑块相邻上调的基因网络提供了更感兴趣的候选治疗靶点(Cell 2020 )
为什么要做单细胞和空间组的关联分析
既然空间组这么好,为什么还需要单细胞的关联分析呢?其实如果做过这两个组学分析的人,应该非常理解,单细胞和空间组的关联分析非常重要,原因就在于,单细胞虽好,但是没有位置信息。空间组虽新,但是精细度令人诟病。
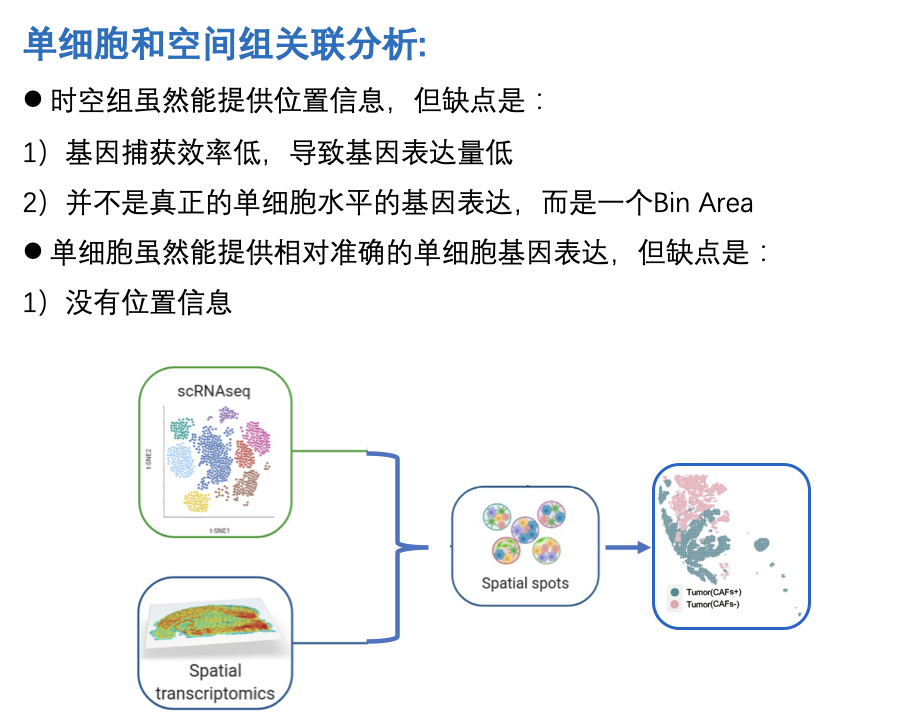
关联分析的路线
作者也提出了,关联分析的步骤应该是,先进行单细胞的细胞类型鉴定,其次是对单细胞的细胞类型映射到空间组中,得到细胞类型在空间芯片上的分布,再去研究组织稳态(例如细胞通讯)。
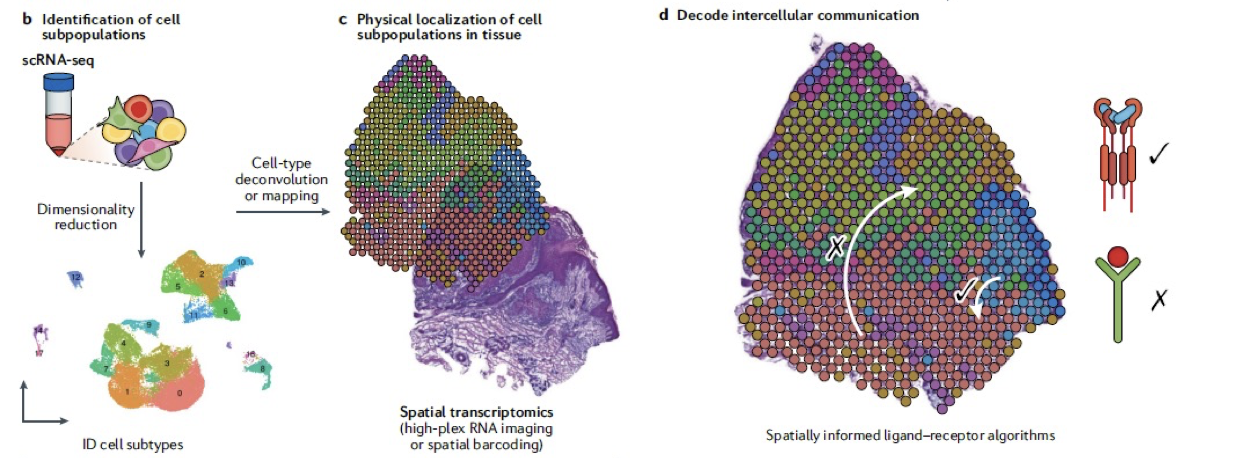
那么问题的关键就比较清晰,如何把单细胞的细胞类型映射在空间芯片上,得到细胞类型的位置关系?
单细胞和空间足的整合分析策略
1. 反卷积🐘
可以通过对来自每个捕获点的mRNA转录本混合物进行解卷积来定位细胞亚群,以预测每个点处细胞混合物中每种细胞类型的比例,评估功效的更方便的策略是通过组合来自多个表征良好的scRNA-seq细胞的转录本测量值来在计算机中创建混合物。
这类方法中比较著名的是SPOTlight,作者也是比较赞赏该软件:
所以我觉得下一篇文章更新SPOTlight的分析教程
2.富集分数🐥
基于scRNA-seq的Spot score 不必局限于对细胞类型进行评分——人们可以对细胞周期期、组织类型、癌性与非癌性以及特异性基因表达方案等特征进行评分。这种评分方法的局限性在于找到由感兴趣的细胞类型表达的真正独特的基因,因为重叠的基因可能会混淆评分。
这类方法中比较著名的有GSVA的Gene Set Variation Analysis函数,Seurat的AddModuleScore函数 、或者采用MIA(multimodal intersection analysis)来进行分析。
MIA 分析在NBT的2020年七月刊上,一篇研究胰腺导管癌的研究性文章《Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas 》一文首次提出,用MIA的方法来做单细胞和空间转录组的关联分析的思路。这个方法我已经在自创的R包Spat(https://github.com/wangjiaxuan666/Spat)上实现,有兴趣的也可以去看看。
3. 映射🐒
这里说的映射特指的思路是:根据找到一些anchor gene,来做样本或者实验质检的批次矫正。这类软件最常用在多个样本的整合中,和批次效应矫正中。最早运用这个技术来做组学技术之间的关联分析应该是Seurat 中10x scRNA和10x ATAC的关联分析中。除了Seurat的整合函数外,还有Harmony ,LIGER等批次校正scRNA-seq批次的软件,也证明了能够在单细胞分辨率空间数据中的成功应用数据整合。
但我总觉这类软件容易存在矫正过枉,但是还是从结果出发,白猫黑猫,逮到耗子就是好猫。
大致原理看起来,LIGER的算法好像高级一点,Harmony最次,但可能有出其不意的效果,Seurat最常用。
-
1️⃣Harmony -> 低纬-> kmeans 聚类
-
2️⃣Seurat -> 低纬-> MNN
-
3️⃣LIGER->NMF
小tips:除了应用在矫正和整合,LIGER和Seurat还能补充未测量基因的表达。Seurat Integration通过使用空间细胞最高评分锚点的scRNA-seq基因表达谱很容易进行插补。得分越高的对在低维空间中具有更多共享的最近邻。相比之下,LIGER和SpaGE117,一种新发表的针对scRNA-seq和单细胞空间数据整合的映射算法,通过获得低维空间中一定数量的相应scRNA-seq最近邻的平均表达进行插补。
注意:这个插补我没用过,不知道具体效果,只能说插补挺新奇的,但是应用很少。
矫正效果的预期
任何软件都是有自己的局限性,有句老话怎么说,再牛逼的生信也解决不了失败的实验🏃♂️🏃♂️🏃♂️。
关联分析也会出现,数据整合效果的问题,这个问题从广义上讲,不匹配可能是由于预测序步骤和/或测序后分析中的错误引起的。此外,当使用独立的scRNA-seq图谱作为参考时,如果其组织条件与新生成的空间数据不能很好的一致,则不匹配可能会加剧。
具体原因可能包括:

好叻,本期完结撒花!😏😏😏
下一篇《SPOTlight分析教程与自己对其中一些源码的改进》预告!