为什么差异基因少, 差异到底是什么?
经常会碰到一些项目,差异基因少,这种情况的原因是非常多因素的。从生信分析的角度上,解决方法最常用的是调整参数。我做了技术支撑也快一年了,在这种问题上也算是有一些心得。
首先找寻问题,要从从下往上一点点寻找才是最保险的。那么差异分析的结果是怎么来的呢
1. 为什么差异基因少
经常会碰到一些项目,差异基因少,这种情况的原因是非常多因素的。从生信分析的角度上,解决方法最常用的是调整参数。我做了技术支撑也快一年了,在这种问题上也算是有一些心得。
首先找寻问题,要从从下往上一点点寻找才是最保险的。那么差异分析的结果是怎么来的呢
从下往上依次是:
- 通过
edgeR或者DESeq2这类软件差异分析而来,筛选标准是|FC| > 2, FDR < 0.05 Tophat2或者Hiseq2这类软件将测序reads比对上参考基因组,并进行定量分析。- 去除核糖体
rRNA - 质控
- 测序
- 测序文库建立
- 实验设计
以上任何一步出现问题都可能导致差异基因少。但是有经验的支撑肯定不会一个个区排查。因为每天都要处理很多项目问题。是不会这样采取一个个追溯寻根的去找项目问题。还是会从最有可能出现问题的地方入手。
想起以前在《意林》还是《读者》看到一篇文章,里面写一个针掉进毛毯中,A会重新模拟角度和力度重新抛一个针去模拟掉落的位置。而B却会把毛毯划分成数个相同大小的区域。一个区域一个区域寻找。我个人肯定觉得A这个方法最快最高效。虽然B的方法最保险,但我所不喜。
2. 一点点寻找原因
2.1 检查样品相关性
简单来说,就是先看样品关系分析结果,例如PCA和样品相关性图。因为组间差异少,很可能和组内的差异大有关。因为从通俗意义上讲。我们关注的是处理导致的差异效应,也就是处理组相比于对照组发生的差异变化。
CK组与T组的差异分析公式:
$$
Treat Effictive = Group Effictve_{(CK-vs-T)} - (Sample Effictive {(CK)} + Sample Effictive{(T)})
$$
注意:上述公式并不是某个算法,只是为了理解而列举出来的概念上的公式
我们可以从空间上去理解,如下图,每个样本有成千上万的基因表达信息,假设所有样本中总共5w个基因组有检测到,那么所有样本在这5w个基因都有自己的定位(表达量)。也就是说样本间的差异距离是体现在N维空间的。同样的从N维上去理解。
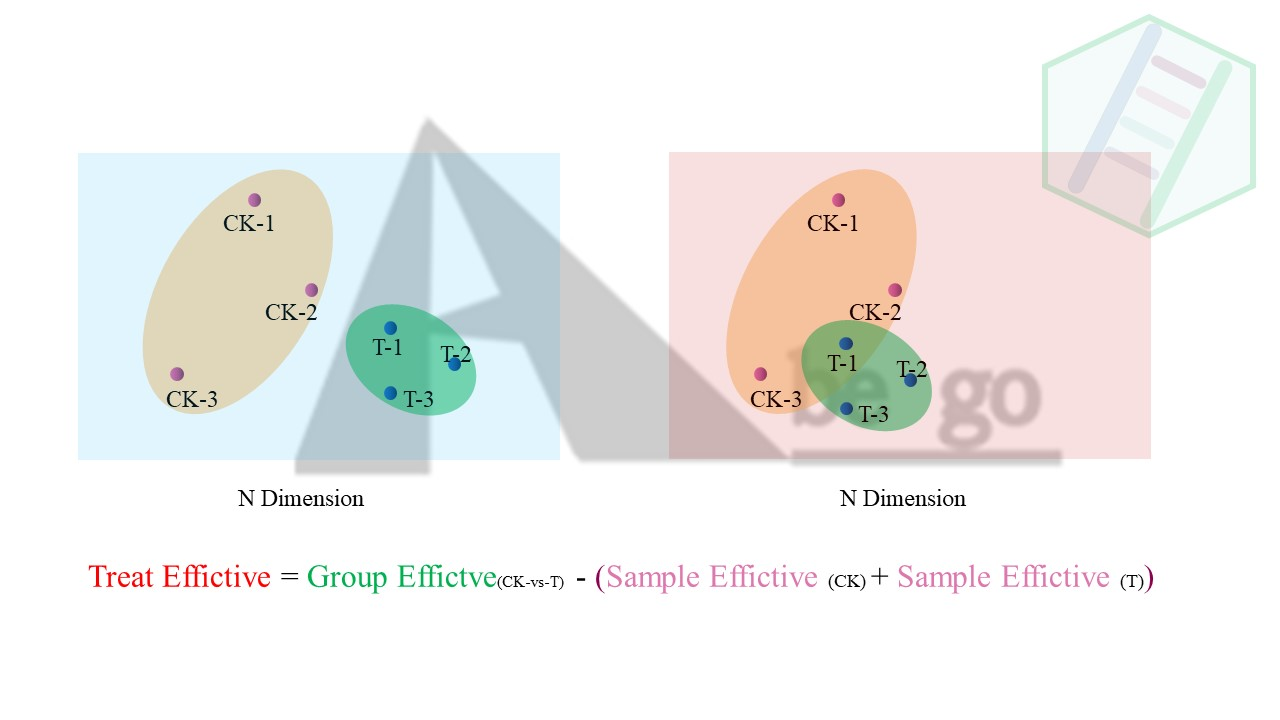
从上述公式我们就可以看到,组内的差异越大,越不利于组间的差异比较。相信这点还是比较好理解的。但是为什么我们要看PCA图呢。或者我们如何根据PCA图等到组间和组内的差异大小呢。
因为PCA(主成分分析)就是通过**重新构建坐标系,从而将高纬数据映射在低纬刻度上。**这样我们就可以通过PC1和PC2的二维空间上近似反应的高纬层面上的数据分布。
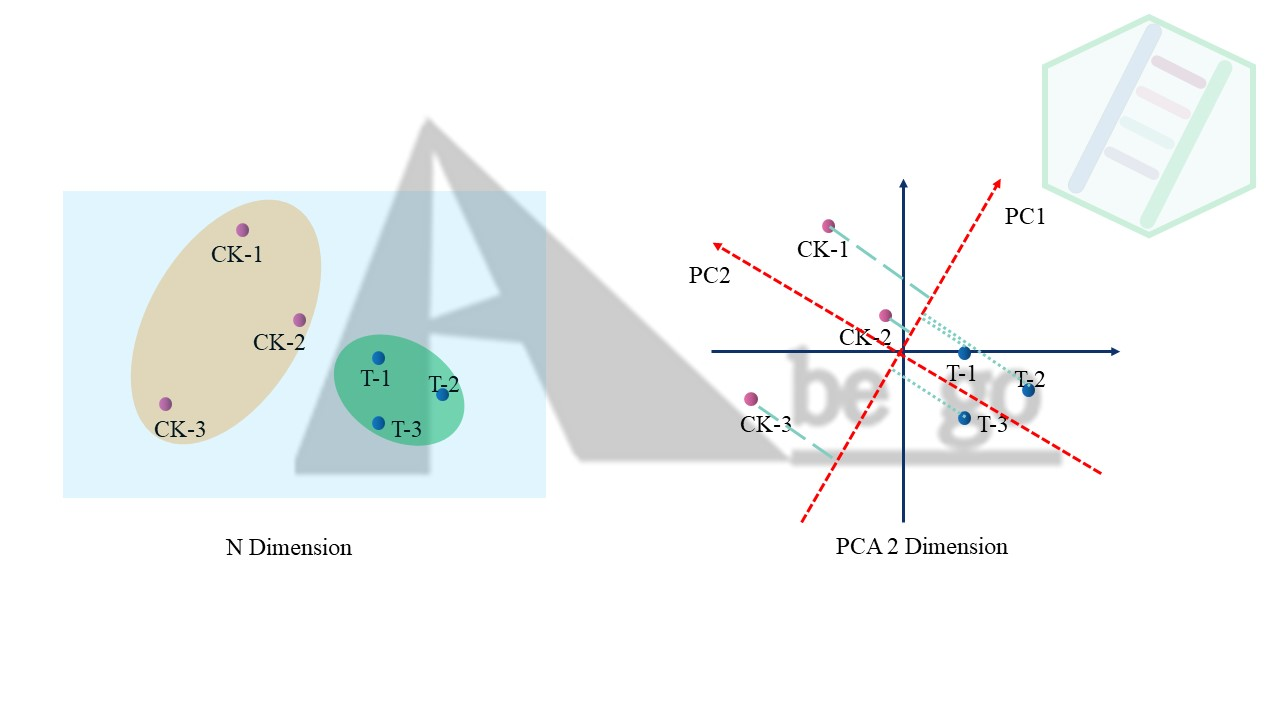
PCA的原理不是本文中的重点,只需要明白PCA能干什么就可以。就不再赘述了。可以看到我们通过降维分析(PCA)将高纬数据映射在PC1和PC2上,那么同样的PCA图就可以反应样品之间的差异。
一个好的分组应该,组内的生物学重复聚在一起,彼此距离相近。
而不好的分组,组与组没有明显的界限,彼此重叠。这种分组差异小,就会导致差异基因少。
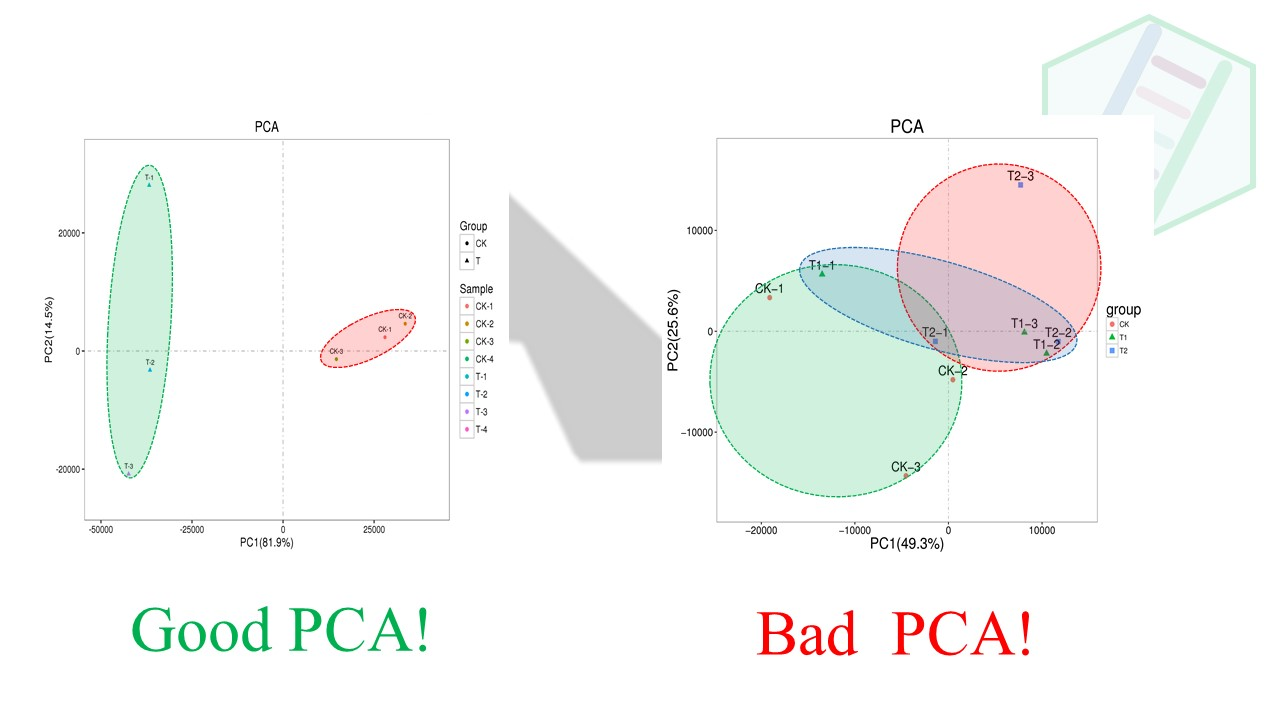
2.2 结尾从头看一篇
目前,各家的流程都是身经百战的,理论上,是不会存在大的Bug。也就以为着分析是很小概率出问题的。虽然上样品相关性可能是最有可能的原因。但是就算我们发现现样品相关性不好,也不要贸然下结论。而是因为从头再把所有结果看一些。
从最开始的数据量、质控,核糖体比率,基因组比率,测序随机性和饱和性,以及最最要的表达量表。都需要进行核对。一个有经验的分析是可以很快的浏览完的。如果这些部分出现问题。依据前者最大的原理,这时候问题的主要的原因就要往前继续推了。就算后面样品相关性不好,也可能是前面这步的问题。
所以还要继续往上走,找测序和建库,参考基因组等原因,去核对为什么基因组比对率低,rRNA含量高等等问题。
但由于这部分实在太多,而且每个项目都不一样,不具有普适性,本文不再继续讨论。本主主要讨论的是,抛去建库测序分析的问题,单单说老师的实验设计导致差异基因少。(这才是项目问题的大头,而且是最难解决的)。毕竟客户自身的问题,你伤不起啊。无尽的扯皮,还不如多想想如何解决问题。
3. 如何解决差异基因少
让我们回到问题的本身,差异基因少,也就是说在所有的基因中满足|FC| > 2, FDR < 0.05的基因少。那么差异倍数|FC|和检验值FDR是什么呢。要回答这个问题。不了解点统计学还真的很难搞。让我们一点点来吧,
3.1 数据分布
在统计学中,数据都是有分布。而且绝大部分还是符合正态分布。何为正态分布。就是极端情况是少见的,而常见的是靠近平均值的数据。啥,不难理解吧。比如人的身高,财富,头发多少,老鼠的体重等等都是符合正态分布的。《中庸》道:
连古人都知道正态分布呢,哈哈,开个玩笑
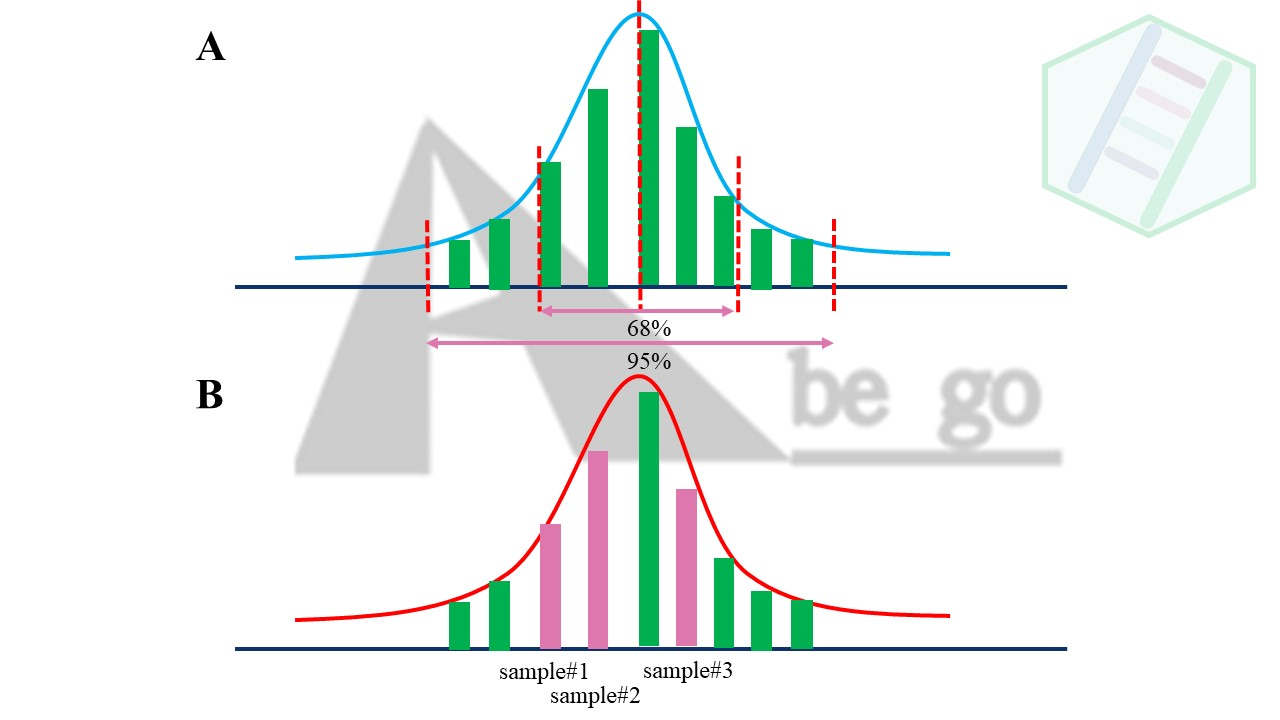
从上述图形,我们可以看出正态分布的大部数值都是接近整体的平均值,而极少数数据是极端大和极端小的。一个标准的正态分布。平均值平均值+-标准差的范围是占整体的68%,而平均值+-标准差*2的范围是占整体的95%。
有没有想起检验值的0.05这个阈值,知道是怎么来的的吧
但是我们在做实验,或者说现实生活中,我们不可能去获得每一个个体的数值,构建正态分布,要不然也用不到统计学了。我们以生物实验为主。我们一般是做三个生物学重复。其实这时候,也是根据三个样本的值计算平均值和方差去估算整体数据的分布范围。
注意:正态分布的积分就是事件发生的概率,平且我们常说的p值也是这个分布的面积相关的。但是p值不是计算自身发生概率。p值还会考虑同等概率事件+极少概率事件。这里不再赘述。因为写起来太多了。
3.2 数据分布的差异
了解了数据分布,那这和我们的两组间的差异分析又有什么关系呢。当然有关系,|FC|我们可以简单理解为差异效应的大小的。但是FDR值又该如何理解。所以我们还是要继续讨论一点统计。
统计上如何确定差异呢?这还是与分布有关系的。简单
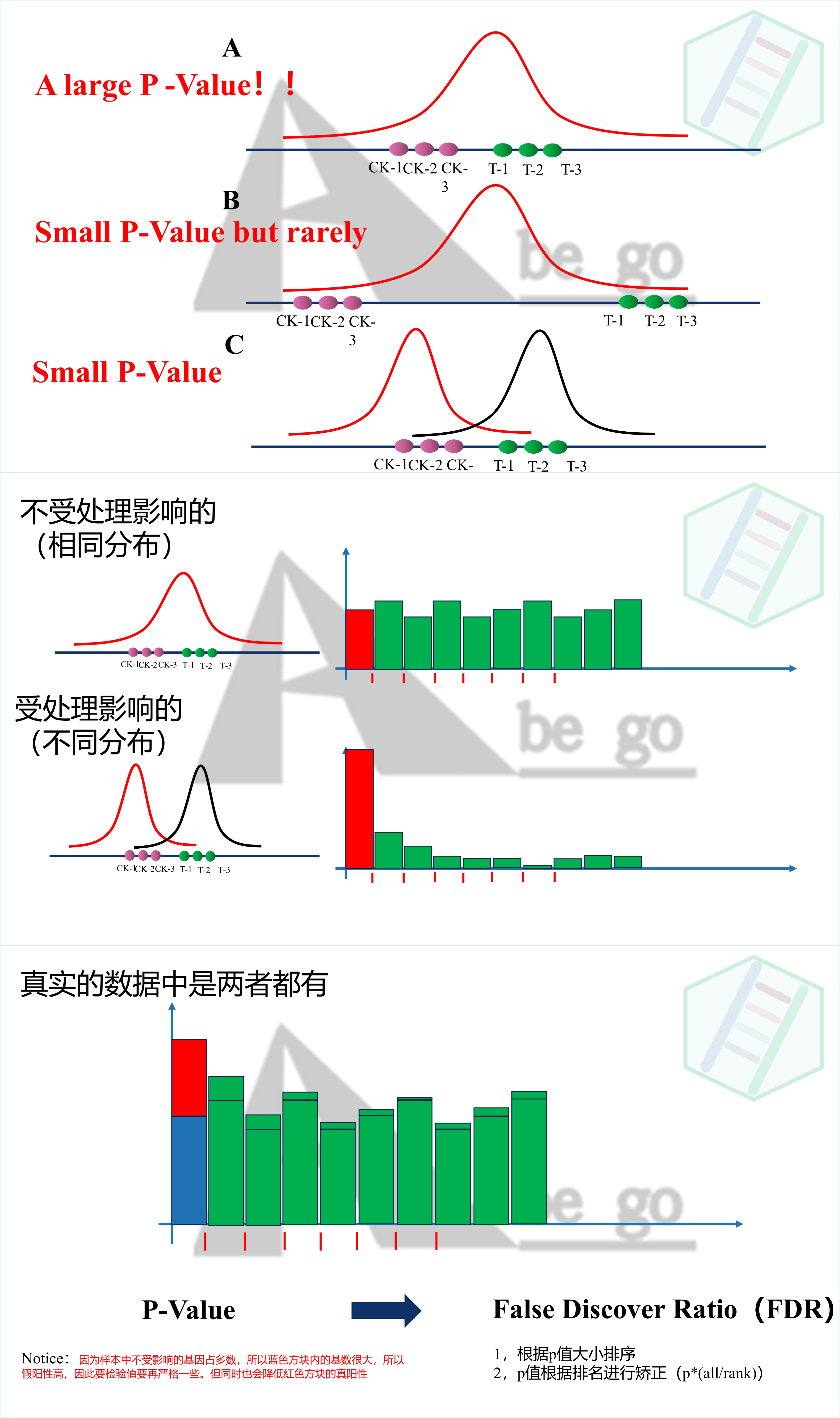
如果数据来自同一个分布,那么p值既有可能会非常大。但是还是存在极少的概率p值是小于0.05的,因为在标准的正态分布中,同一个分布随机抽取样本进行检验,p值是平均分配的。
但是如果样本是来自两个不同的数据分布,p值就会非常小。这才是我们认为真正处理所导致的差异。
但是很明显我们关注的对照组CK与处理组T组间的所有基因,绝大部分是不受我们处理影响,一部分基因是收到我们处理的影响的,那么两组之间的基因进行差异比较的时候,既有来自一个数据分布的(不受处理影响)也有一部分是两种分布(收到处理影响的)。
4. 影响差异的因素
从上面,我们可以得知,从统计上如何理解差异比较。那么回到我们的主题上,为什么我们的差异基因会少呢。|FC|是平均值的比较,检验值FDR是数据分布的统计检验。还是回到统计学上理解。
两组间差异分析常用的检验是T-test。
概念公式:
$$ P = \frac{信号}{噪音} = \frac{DifferentBetweenGroupMeans}{Variabity Of Groups} $$
所以很明显我们可以看到检验p值以及根据p值而来的FDR值也是考虑FC的,只是这里的考虑是为了验证是否两组间有差异,并没有|FC|多大是变化,多小时没有变化的这个概念。所以后续差异分析还是考虑|FC| > 2的。
值得探究的是检验计算的分母Variabity Of Groups,这个概念和数据分布有很大的关系。
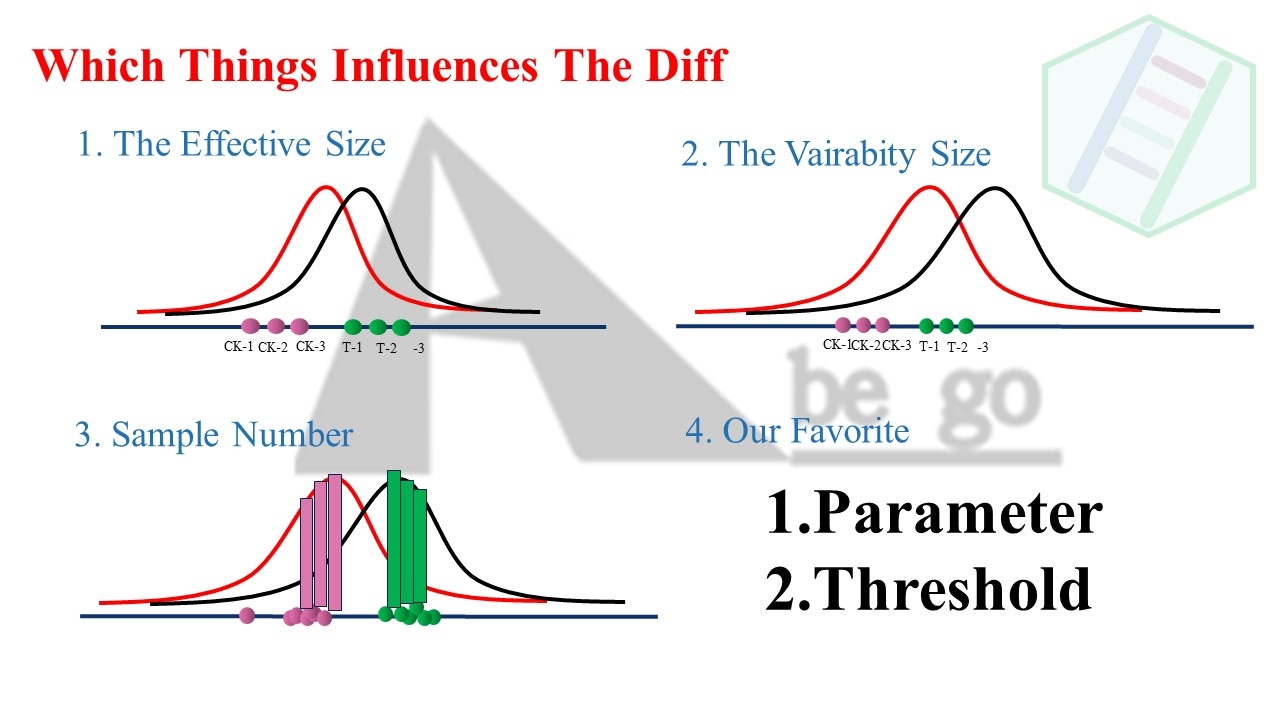
简单来讲:
- 处理因素的强度和差异分析的效果最为相关,这里的因素包括实验设计内的和实验设计外的。很多老师反馈自己的实验设计的强度时适合的,但是并没有考虑那些实验设计以外的因素,比如物种差异,生物的稳态调节等等。
- 还有就是数据的变异程度,如果组内相关性不好,组内差异大,就会导致三个样本预估的数据分布呈现一个非常宽的分布范围。这对于差异分析的检验来说时非常不利的。
- 另外就是样本数了,根据中心回归原则,样本数越大,越接近数据的真实分布,会让数据分布的范围缩小,减少因为预估而导致对分布估算过大。这也是
16s等微生物、生态实验设计中,样本数多的原因。因为微生物群落不稳定,受因素影响多。而样本数目多,可以很好的预估数据的真实分布范围。 - 当然,我们最喜欢的就是调整参数。把
FDR< 0.05换成P< 0.05,或者调整阈值等等。这往往来的最粗暴最简单。
5. 如何避免差异基因少
这是很高深的领域,首先如果有预实验或者类似实验设计的结果。都可以很好的帮助我们进行评估差异基因的数目。但是如果没有,良好的预实验可以帮忙避免此类问题,但是对于缺乏预实验,并且已经拿到测序数据的结果。
可能更多的是如何矫正结果。不妨看看我的R包fixbatch.这个R包汇集了sva,snm以及中位数矫正的思路,可以尽量剔除无关的批次效应(背景噪音),聚焦于我们关注的生物学问题上。
但是这个R包缺乏维护,有问题可以在这里留言,我会尽量完善。