生信软件学习-CellRanger的安装和使用
最近因为换了工作,真正的做了一名分析💻,但是因为时间紧任务重,所以一直都是用自己自学的R来支撑工作。但这终究不是个办法。所以除了工作内的分析单细胞和空转的内容外,我大概也有自己的计划📕:
- 第一就是学习安装和使用自己之前了解但没实际操作过的重要软件🔬
- 再学一门流程语言,之前的同事建议我学个python或者perl🏇
先说学习软件吧,首先是单细胞分析的cellranger,因为下游分析我基本上都用R可以解决,所以主要还是上游分析的cellranger的熟悉和使用。
安装可以参考官网的介绍, 软件也可以从官网下载。值得注意的是:
🅾️🅾️🅾️ 一定不要选择从网页上下载,那个是有问题的,说多的都是泪,一定要要从crul或者wget方式下载。
当下载好了以后,cd到下载目录下,用tar解压缩后,就已经安装好了(免安装版本?!!),然后记得要导入环境变量中,要不然以后使用只能用绝对路径了。像我就是用的centos7的集群,我就是在home下vi ~/.bashrc,然后手动修改的,然后source ~/.bashrc就搞定了。
cd /download file
tar -xzvf cellranger-6.0.1.tar.gz
# 添加环境变量,目录要换成解压后的文件目录!
export PATH=/opt/cellranger-6.0.1:$PATH
当解压并导入环境变量后,我们可以检查下是否安装成功。在终端输入
cellranger testrun --id=tiny
这样Cellranger会自己进行自检查,但是这个用时还蛮久的。总之最后会生成一个tiny.mri.tgz检查报告文件,自己也可以解压出来看看。不过我感觉直接在终端输入cellranger count,只要出来的结果不是command not found应该就是安装成功了。
分析数据
如果是一个建库样本,无论测多少次,都可以直接用cellranger count来做分析。但如果是不同的建库样本,想要放在一起分析就需要(疑问:谁会这样做):
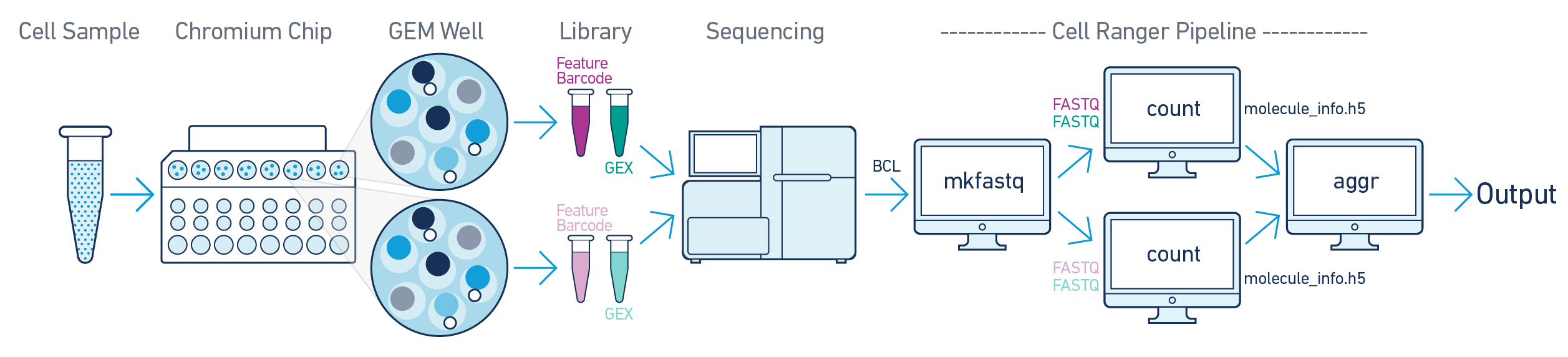
先用cellranger count分别定量,然后用cellranger aggr合并样本。
但是随后我发现,cellranger aggr,的功能,好像Seurat也可以实现啊,这里面就纠结了,到底是选用官方的cellranger aggr,还是选用分析常用的Seurat。甚至我还特意跑去了网上搜索了下。根据Seurat的团队所述,其并不推荐cellranger aggr,原因是该方法可能会丢失大量的数据[1]。但也有人认为这取决于实验之间的批次效应,以及预期结果,简而言之就是如果有很大的批次效应,可以尝试用cellranger aggr做做看[2]。
我肯定选择Seurat,额,毕竟这样才能愉快放在一起分析
Cellranger count分析数据
首先要现在相当多的数据:
- 1)基因组文件
- 2)测序数据
下载基因组文件
human GRCH38 (大小11G🍵,这个文件不仅有fa和gtf文件,而且star的指引文件都构建好了。而且10x公司针对自己的产品做了部分优化)[3]
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz
mouse mm10 大于10G大小🍵
curl -O https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz
下载测序文件
接下来,从 10X 官网上的一个公开数据集下载 FASTQ 文件。本示例使用来自人类外周血单核细胞 (PBMC) 的1000 个 细胞 数据集,这些数据集由淋巴细胞(T 细胞、B 细胞和 NK 杀伤)和单核细胞组成🎯🎯🎯。
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_fastqs.tar
tar -xvf pbmc_1k_v3_fastqs.tar
定量分析
我们需要用到cellranger count命令来进行定量分析,可以先看命令的帮助文档,帮助我们理解其参数的含义。
cellranger count --help
### output start
#嗯,自己看吧,我懒得写了
### output end
总之,我用的参数是
cellranger count --no-bam\ #不需要bam文件
--nosecondary\ # 不需要它分析聚类和细胞分群
--disable-ui\ # 不需要web展示
--id=run_count_1kpbmcs \ #分析名称
--fastqs=/pbmc_1k_v3_fastqs \ #fq文件目录
--sample=pbmc_1k_v3 \ #样本名称
--transcriptome=/refdata-gex-GRCh38-2020-A #刚刚下载的基因组文件
结果展示
一般这1000个细胞,可能4核16G能够在8个小时内结束,还是很快的,结果乱起八糟的,其中有用的是outs文件下面的文件:
drwxr-sr-x 2 4.0K Apr 12 21:50 filtered_feature_bc_matrix
-rw-r--r-- 1 8.6M Apr 12 21:50 filtered_feature_bc_matrix.h5
-rw-r--r-- 1 651 Apr 12 21:57 metrics_summary.csv
-rw-r--r-- 1 87M Apr 12 21:51 molecule_info.h5
drwxr-sr-x 2 4.0K Apr 12 21:47 raw_feature_bc_matrix
-rw-r--r-- 1 17M Apr 12 21:47 raw_feature_bc_matrix.h5
-rw-r--r-- 1 2.4M Apr 12 21:57 web_summary.html
明显的filtered_feature_bc_matrix文件下的是可以直接作为Seurat的输入文件路径的(就是这么方便!)。
好了、完结!讲真,成熟的、或者商业化软件真是方便的很,反观一些发了文章就不管的软件都长毛了,也不维护下,给旁人徒增烦恼~,不过也强求不得。
参考文献:
[1] [Cell Ranger “Aggr” read-depth normalization vs. Seurat NormalizeData](https://github.com/satijalab/seurat/issues/672)
[2] [Cellranger aggr versus Seurat](https://www.biostars.org/p/433937/)
[3] [Build Notes for Reference Packages](https://support.10xgenomics.com/single-cell-gene-expression/software/release-notes/build#mm10_#{files.refdata_mm10.version})