膜蛋白的分析由于它们的疏水性,复杂的翻译后修饰(PTM)以及它们以低丰度,一直以来都是蛋白研究的难点。
膜蛋白富集实验方法
[TOC]
细胞膜蛋白
膜蛋白是一类与生物膜相互作用或从属于生物膜的蛋白质。膜蛋白可根据其位置和与膜的相互作用分为三个部分:integral (跨膜蛋白); peripheral(外周蛋白); 或lipid-anchored(脂质锚定蛋白)[1]。
- 外周膜蛋白以几种方式附着在膜的一侧,如平面内α-螺旋。
- 跨膜蛋白跨越脂质双层并且是两亲性的。它们的亲水区域突出到细胞质或细胞外环境中以与可溶性蛋白质和分子相互作用,而疏水区域用于将蛋白质嵌入脂质双层中。
- 而脂质锚定蛋白确实直接结合在磷脂双子层上
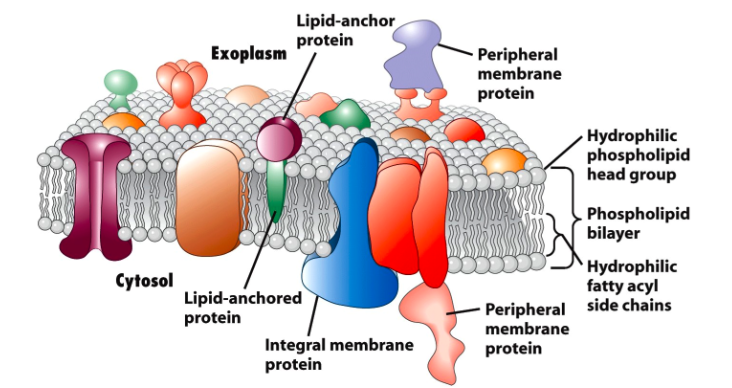
膜蛋白在各种细胞过程中起重要作用,例如细胞粘附,免疫应答,代谢和信号转导。它们可以作为转运蛋白,受体和结构蛋白。因此,膜蛋白是蛋白质组学研究的热门目标,也是药物开发的常用候选药物。然而,膜蛋白具有低丰度,溶解度低,以及酶切位点可及性低等特性,也就限制膜蛋白研究。
质谱蛋白质组学方法减轻了膜蛋白鉴定的难度。然而膜蛋白的研究困难并不是消失了,就目前而言,膜蛋白质组分析的主要困难还是主要集中在于膜蛋白的富集上。因此膜蛋白的研究手段一般第一步就是膜蛋白的富集。
膜蛋白的富集
由于整合膜蛋白质含量低,因此膜蛋白的富集对于蛋白质组学分析至关重要。有几种策略[2]:
- 亚细胞分级(使用甘油或山梨糖醇进行梯度密度离心梯度收集细胞膜
- 表面活性剂破坏细胞膜膜,
- 然后使用氯仿/甲醇溶液进行脱脂以提取和溶解膜来自脂双层的蛋白质,
- 跑PAGE胶分离膜蛋白, 还有人用抗体和生物素亲和纯化, 也有用高盐和高pH去除非膜蛋白。
- 蛋白酶消化(胰蛋白酶和蛋白酶K)
- 质谱和比对计算分析
文献推荐是用蛋白酶K去切割,因为膜蛋白缺乏胰蛋白酶位点,所以部分膜蛋白可能切割不彻底,从而保留大分子结构,被质谱过滤掉。应该用广谱蛋白酶K,但是我也看其他文献就是直接用胰蛋白酶去切割蛋白获得肽段。具体那个参考文献忘了,文末可以找找。
膜蛋白鉴定为其生物过程的调节提供了有价值的见解。具有更高质量准确度的先进仪器将允许更高的蛋白质组覆盖,这将加速膜蛋白的分析。这里推荐DIA蛋白检测技术。
因为DIA是数据采集非依赖模式,不会想Labelfree每个窗口选择丰度最高的top10或者top20的碎片去打质谱。而是全部去打质谱。其样本的重复性和检出率都是比蛋白质谱其他技术要好。
具体的实验步骤,我们推荐老师可以参考如下文献。虽然也有文献用试剂盒进行提取,很多公司都有对应的试剂盒产品,但一般都是针对动物的。如果植物想做的话,可能还要提取原生质体。
注意:下列方法可能比较复杂,如果不做膜蛋白富集也可以直接送组织样到测序平台测序,但是有两点:
- 量要大,因为膜蛋白丰度低,容易检测不到
- 不保证一定检测到绝大部分的膜蛋白,甚至检测到多少膜蛋白也是不好确定的。
动物文献参考

这篇文章是研究鹿茸再生干细胞的膜蛋白。根据文献报告膜蛋白的富集方法:
- 使用Minute TM质膜蛋白分离试剂盒(Invent biotechnologies,Eden Prairie,MN,USA)按照制造商的程序分离细胞膜蛋白。
- 使用无菌细胞刮刀收获每个样品的组织到三个100-mm培养皿(约300w细胞),
- 并用冷磷酸PBS洗涤两次。用含有蛋白酶抑制剂的缓冲液A重悬浮细胞沉淀,并进行10个超声处理(5秒/ 5秒关闭)。细胞悬浮液转移到过滤器滤芯,并在16000离心克 30秒。细胞沉淀再次重悬,细胞核(700× g,1分钟),胞质组分和膜蛋白(16,000× g)通过梯度离心连续分离,30分钟)。将总膜蛋白质用缓冲液B重悬,细胞器膜(7800×g,20分钟)和质膜(16,000× g,20分钟)通过梯度离心再分离一次。将质膜蛋白沉淀溶解于200μL蛋白质提取缓冲液中
植物文献参考

这篇文章[3]是研究拟南芥叶细胞的膜蛋白。:
植物材料
拟南芥生态型Columbia-0在22℃下在土壤上生长,短日照(9小时光照/ 15小时黑暗,170μE)和70%相对湿度。6-8周后收获绿叶(包括叶柄)并用于质膜分离。
质膜隔离(不翻译了,怕错)
About 100 g of plant material was homogenized, using a knife blender, in 150 ml of 330 mM sucrose, 50 mM MOPS-KOH, pH 7.5, 5 mM EDTA, 0.2% casein hydrolysate, 0.6% polyvinylpolypyrrolidone (PVPP), 5 mM ascorbate, 5 mM DTT. PVPP, ascorbate and DTT were added immediately before use. Immediately after homogenization, PMSF was added to a final concentration of 0.5 mM together with 1.5 ml of a protease inhibitor cocktail for plant cell and tissue extracts (P9599; Sigma, St. Louis, MO, U.S.A.) in DMSO. The homogenate was filtered through a 200 µm nylon mesh and centrifuged at 10,000×g for 15 min; the supernatant was saved and centrifuged at 30,000×g for 50 min. The resulting microsomal pellet was resuspended in 6 ml of resuspension medium: 330 mM sucrose, 5 mM K-phosphate, pH 7.8, 0.1 mM EDTA, 1 mM DTT (freshly added) and 50 µl protease inhibitor cocktail. The resuspended membranes (6.0 ml) were added to an 18.0 g phase mixture to produce a 24.0 g aqueous polymer two-phase system with a final composition of 6.1% (w/w) Dextran 500, 6.1% (w/w) PEG 3350, 5 mM K-phosphate, pH 7.8, and 3 mM KCl. Plasma membranes were then purified by aqueous polymer two-phase partitioning as described previously (see Larsson et al. 1994, for the most recent update). The final upper phases were diluted at least two-fold with 330 mM sucrose, 5 mM K-phosphate, pH 7.8, 0.1 mM EDTA, and plasma membranes were pelleted by centrifugation at 100,000×g for 1 h. The whole preparation procedure was performed at 4°C. The plasma membrane pellet, containing 4–6 mg of protein, was resuspended in 0.5 ml of resuspension medium and 5 µl protease inhibitor cocktail, and stored in liquid nitrogen until used. Protein concentration was determined according to Bearden (1978) with BSA as standard.
The plasma membrane vesicles, which are largely cytoplasmic-side-in, were turned inside-out by treatment with the detergent Brij 58 (Johansson et al. 1995) to remove soluble proteins enclosed in the vesicles as well as loosely bound, contaminating proteins. This was done by mixing, at room temperature, stock solutions of 2 M KCl and of 2% (w/v) Brij 58 in 330 mM sucrose, 5 mM potassium phosphate, pH 7.8, with plasma membranes to give a detergent to protein ratio of 10 : 1 (w/w) and a KCl concentration of 0.2 M. Since this treatment also results in smaller vesicles (Johansson et al. 1995), the final centrifugation was run for 2 h at 100,000×g to increase recovery, which was 50–60% on a protein basis. The final plasma membrane pellet was resupended in half the original volume (usually 250 µl) of resuspension medium and 2.5 µl protease inhibitor cocktail, and used for SDS-PAGE.
SDS-PAGE胶分离
SDS-PAGE was performed essentially according to Laemmli (1970) using 12 cm gradient (12–20% acrylamide) separation gels. Gels were run at 4°C for 1 h at 5 mA followed by 18 h at 8 mA. Protein was stained with Coomassie Brilliant Blue R 250.
胰蛋白酶消化和labelfree 检测
- 切割出可检测到的高于15kDa的30个蛋白质条带,
- 液氮速度,-80度保存,干冰运送即可,这点可以和我们当地的销售联系
细菌膜蛋白的提取
在革兰氏阴性菌的表层,有由肽葡聚糖形成的细胞壁,在壁的外面,又有由蛋白质、磷脂质、脂多糖形成的膜层,与里面的细胞质膜相对应,特称此层为细菌外膜。外膜比细胞质膜的磷脂质含量低,但脂多糖的含量则比较高。外膜的蛋白质与细胞质膜不同,主要部分为数种蛋白质所构成。其主要蛋白质的部分与特异的内面的肽葡聚糖以共价键结合。脂多糖存在于外膜的最外层。在外膜中仅知有磷脂酶,在细胞与外界的联系中,已看到有许多功能的蛋白质,即有各种噬菌体、维生素 B12、大肠杆菌素等的受体存在,而其一部分蛋白质则与 DNA 的复制、细胞分裂有关,此外,外膜对水溶性低分子物质容易透过,但对抗菌物质则是透过的屏障,它对革兰氏阴性菌间的透过性带来很大的差异。
溶菌酶能有效地水解细菌细胞壁的肽聚糖,其水解位点是 N-乙酰胞壁酸(NAM)的 1 位碳原子和N-乙酰葡萄糖胺(NAG)的4 位碳原子间的β-1.4 糖苷键。
预实验细化版本
- 洗涤菌液:取 1ml 菌液,4 度,2600g 离心 10min 去除上清,再用预冷的 PBS 溶液重悬,混旋 30s,2600g 离心 10min,去除上清。(预先读取离心管质量,再秤出带沉淀离心管质量,相减获得菌体质量。按 1.5g 菌 10 mL 的比例加入 20%(200g/L)的蔗糖溶液,混悬 30s, 2600 g 离心 10min 弃除上清。
以下所有涉及的溶液体积均针对 1.5g 菌样,具体的体积需根据实际菌量换算
- 外膜裂解和分离:沉淀中加入 18 mL 预冷的 20% (200g/L)蔗糖溶液,混悬均匀,再缓慢 滴入 9 mL 2M 蔗糖(68436g/L), 10 mL 0.1M Tris-Hcl (pH 7.8), 0.8mL 1% EDTA-Na, 1.8mL 1% 溶菌酶,37 度 温育 30min 溶液粘稠后,加入 DNase 至 3ug/mL,37 度温育 30min-1hr。
这一步主要是为了水解细胞壁,分离原生质体和细胞壁以及外膜,有显微镜的话,可以取一滴观察酶解过程中的结构变化,大部分细胞壁被水解只有卵状物存在后就可以停止酶解
-
分离原生质体与外膜:30 度下 5200g 离心 15min。清中包含的粗的外膜(OM),60000g 4 度 离心 1h 得到外膜沉淀,保存于-20 度。
-
沉淀为原生质体,加入 5mL 0.05M PBS 蛋白酶抑制剂, 超声 2min (超 5s,停 10s,总超声时间 6min)后 30 度下 5200g 离心 20min。上清中含粗内膜(IM),用 35ml 100mM碳酸钠稀释后 4 度下混悬 1hr 分离可溶和不可溶的成分,4 度下 60000g 离心一小时,沉淀为内膜。
- 膜蛋白提取:2% SDS 超声下冰浴提取蛋白(体积比 1:10 左右加裂解液,同时加蛋白酶抑制剂),超声 20min (超 5s,停 10s) 后 4 度下 15000g 离心 30min,上清即为膜蛋白提取物
参考文献
[1] Lodish H. Molecular cell biology[M]. Macmillan, 2008.
[2] Gilmore J M, Washburn M P. Advances in shotgun proteomics and the analysis of membrane proteomes. Journal of* proteomics*, 2010, 73(11): 2078-2091.
[3] Alexandersson, E., Saalbach, G., Larsson, C., & Kjellbom, P. (2004). Arabidopsis Plasma Membrane Proteomics Identifies Components of Transport, Signal Transduction and Membrane Trafficking. Plant and Cell Physiology, 45(11), 1543–1556. https://doi.org/10.1093/pcp/pch209
[4] Arnott, D., Kishiyama, A., Luis, E. A., Ludlum, S. G., Marsters, J. C., & Stults, J. T. (2002). Selective Detection of Membrane Proteins Without Antibodies: A Mass Spectrometric Version of the Western Blot. Molecular & Cellular Proteomics, 1(2), 148–156. https://doi.org/10.1074/mcp.M100027-MCP200
[5] Komatsu S., Konishi H., & Hashimoto M. (2007). The proteomics of plant cell membranes. Journal of Experimental Botany, 58(1), 103–112. https://doi.org/10.1093/jxb/erj209
[6] Membrane protein identification by shotgun proteomics – Creative Proteomics Blog. (n.d.). Retrieved 8 September 2019, from https://www.creative-proteomics.com/blog/index.php/membrane-protein-identification-by-shotgun-proteomics/
[7] Proteomic-analysis. (n.d.). Retrieved 8 September 2019, from Sigma-Aldrich website: https://www.sigmaaldrich.com/china-mainland/zh/technical-documents/protocols/biology/purifying-challenging-proteins/proteomic-analysis.html
[8] Savas, J. N., Stein, B. D., Wu, C. C., & Yates, J. R. (2011). Mass spectrometry accelerates membrane protein analysis. Trends in Biochemical Sciences, 36(7), 388–396. https://doi.org/10.1016/j.tibs.2011.04.005
[9] Wang, D., Ba, H., Li, C., Zhao, Q., & Li, C. (2018). Proteomic Analysis of Plasma Membrane Proteins of Antler Stem Cells Using Label-Free LC–MS/MS. International Journal of Molecular Sciences, 19(11), 3477. https://doi.org/10.3390/ijms19113477