ggtree是Y叔的,也是微信公众号biobabble的号主,称号呢,挺多的。简而言之就是国内R开发者中的佼佼者。尤其擅长的是可视化。也就是我们比较关注的绘图部分。
ggtree简介
ggtree是Y叔的,也是微信公众号biobabble的号主,称号呢,挺多的。简而言之就是国内R开发者中的佼佼者。尤其擅长的是可视化。也就是我们比较关注的绘图部分。
之前我们介绍的clusterprofile就是他的作品,不过除此以外,他还有一个非常优秀的R包就是进化树的可视化R包tree,注意是
其实我最早之所以知道Y叔就是因为ggtree,但是也是我了解很晚的一个R包,原因就是因为,我没有树文件,让我分析作为个模板,工作和生活中都没有很好的应用场景。
但是最近因为办生信培训班,知道了ape这个包里有写出树文件的功能,所以这样的话,素材就是有了。所以也是一点点再看。
其实本文也有关于如何导出和导入热图的聚类树的[教程](# section01)
进化树背景知识
我对聚类树和进化树的结果倒是会看,但是树文件就很不友好了。基本上是一团懵逼。尤其是自己对进化树其实没什么深入了解。
所以一些基本知识的欠缺导致入门的时候,经常不明白这个node是什么鬼东西。
进化树很好理解,就是两点之间的
除此以外,还有进化尺度的衡量,树枝🌿越长代表关系越远,这个其实也是行走的距离的标准之一。
除此以外,还有某些进化上时间的关系,这个时候就需要一个标准,一个已知进化时间的外群,或者已知进化时间的样本,就可以根据这个标准的时间,去计算其他样本的进化时间。
ggtree的实操
第一步,当然是要得到树文件🌿,进化树也大致分为两种,一种是
d <- dist(data)#计算样品间的距离
h <- hclust(d)#转换成聚类树的格式
另外一种就是根据核苷酸的替代和变化,构建的进化树,可以用MEGA X,也可以使用在线网站制作。这种网站很多。尤其是就职的公司公众号,发布了不少这方面的教程。就不在自述了。
ggtree的操作文件
这次教程的示例来自R自带的数据包mtcars,这次够友好吧,终于不拿公司的流程中的表达量总表来当例子了
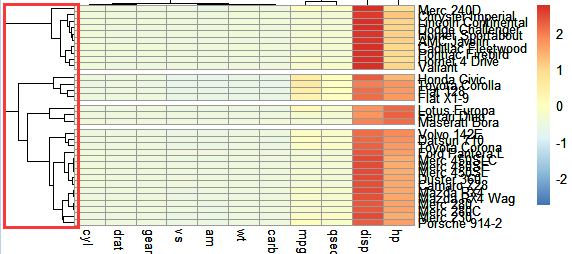
用热图聚类的结果当作输入文件 {# section01}
将聚类热图的结果导出文件,这里可是能导出pheatmap的聚类结果奥。并且按照聚类的结果新增了group这一列,代表基因的分类情况。
path = "C:/Users/woney/Desktop"
setwd(path)
require(pheatmap)
p1 <- pheatmap(mtcars,scale = "row",cutree_rows = 4)
cut <- cutree(p1$tree_row,k=4)
dat_cut <- cbind(mtcars,"group" = cut)
write.table(dat_cut,"heatmap_cut_id.xls",sep = "\t")
将热图的聚类书读出
接下来,就要根据热图的聚类结果,导出聚类树🍀,如图
安装ggtree
if(!requireNamespace(“BiocManager”,quietly = TRUE))
install.packages( “BiocManager”)
BiocManager :: install(“ggtree”,version =“3.8”)
browseVignettes( “ggtree”)#打开帮助文档
读取数据
require(ape)
hc <- p1$tree_row
write.tree(as.phylo(hc),file = "mytree.nwk")
这样我们的桌面上就多了一个
将树文件导入R
用read.tree函数导入
require(ggtree)
inname <- "my_tree.nwk"
tree <- read.tree(inname)
ggtree(tree) + geom_tiplab()#这个文本没有打印完全,这个可以设置par全局画布的大小来改变,但是这里就不展开了,反正是栗子
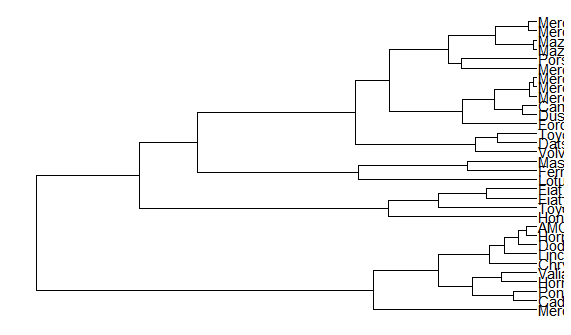
到此,我们已经有了树以及树文件。接下来就是ggtree登场的时间了.
ggtree的树图美化
和大部分图形一样,分为两种
- 全局美化
- 细节美化
因为ggtree的功能实在是太强大了,另外因为作者是中国人,所以基本可以说,只要你敢想,没有做不到的,实在不行去知识星球找作者Y叔,答疑、提要求等等,很NICE。
帮助文档的学习
所以这里,我只是简单论述一下,我学习ggtree遇到的一些障碍和心得体会。所以只是从大概上说一说。如果各位有兴趣。大可以在谷歌😆 上搜索一下。或者从上文中寻找打开帮助文档的命令。
帮助文档:
https://bioconductor.org/packages/release/bioc/html/ggtree.html
全局美化
layout布局美化
首先最重要的当然是树图的layout了。
ggtree(tree,layout = "circular")
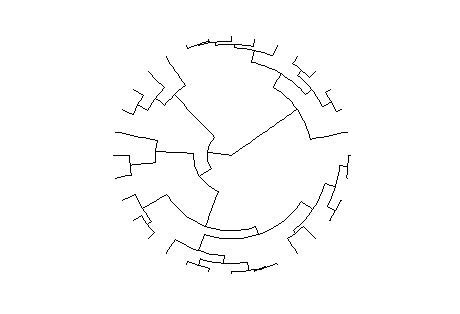
其中ggtree设置了很多布局参数。如下图:

各位可以随心所欲的调整。😜
ggplot2的图形美化参数
ggtree(tree) + scale_x_reverse()#将x轴反过来
ggtree(tree) + coord_flip()#将x轴和y轴置换
ggtree(tree) + coord_polar(theta='y')#转换成极坐标,就是所谓的方图变圆图
ggtree(tree) + xlim(-10, NA)#坐标轴限制某一范围内
各位可以试一试以上参数,会有更加直观的了解。那么除此以外了,ggtree是非常高水平的继承了ggplot2的思想和语法以及有优点。这就是说如果你了解ggplot2就会非常了解ggtree。甚至利用ggplot2来美化ggtree。
比如
用theme主题参数和scale参数来修改坐标轴,名称,以及主题。甚至也能用facet参数来进行分面。
ggtree自带的geom图层美化
以下就是ggtree自带的图层(我删去了一部分,完整的看帮助文档),还是需要什么了解什么吧。
| 层 | 描述 |
|---|---|
| geom_cladelabel | 使用条形和文本标签注释分支 |
| geom_hilight | 突出显示带矩形的分支 |
| geom_label2 | geom_label的修改版本,支持子集 |
| geom_nodelab | 节点标签的图层,可以是文本或图像 |
| geom_nodepoint | 使用符号点注释内部节点 |
| geom_point2 | geom_point的修改版本,支持子集 |
| geom_rootpoint | 用符号点注释根节点 |
| geom_text2 | geom_text的修改版本,支持子集 |
| geom_tiplab | 尖端标签层,可以是文本或图像 |
| geom_tippoint | 用符号点注释外部节点 |
| geom_tree | 树结构层,支持多种布局 |
我个人比较感兴趣的
geom_cladelabel,geom_hilight,geom_tiplab,这些geom图层。所以主要还是讲这些。
显示树尺度(纯copy帮助文档)
要显示树比例,用户可以使用geom_treescale()图层。
ggtree(tree) + geom_treescale()

显示节点/提示(纯copy帮助文档)
显示所有树的内部节点和提示,😉可以通过使用添加点的层来完成geom_nodepoint,geom_tippoint或者geom_point。
ggtree(tree) + geom_point(aes(shape=isTip, color=isTip), size=3)

显示标签(纯copy帮助文档)
用户可以同时使用geom_text或geom_label显示节点(如果可用)和提示标签,或geom_tiplab仅显示提示标签:
p + geom_tiplab(size=3, color="purple")

细节美化
进化树的细节美化主要依靠的是节点(node)这个参数,这个参数也是我之前已知没有理解的点。后来才发现这个node其实就是进化树上的分枝点。
如下图:
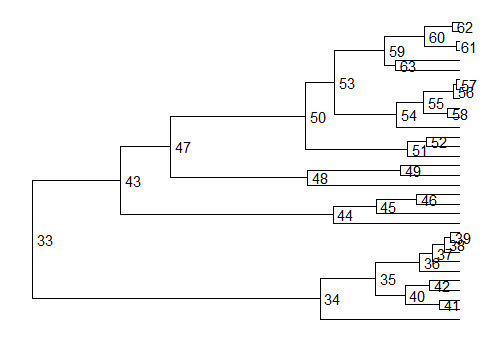
这样我们知道了node值就可以知道我们修改那些进化树的分支。
获取NODE参数
所以在进行细节上的美化之前,我们一定要知道所需要美化的分枝所在的位置(node)。获得node方法:
- 用以下命令绘制图形,看图形得知node
ggtree(tree) + geom_text2(aes(subset=!isTip, label=node), hjust=-.3) + geom_tiplab()
- 用以下命令得知,所关注的node位置,毕竟样本太多的时候,眼睛是看不过来的。
MRCA(tree, tip=c('A', 'E'))#这里的A和E就是所关注的样本名称
- 用
viewClade查看分枝
viewClade(p+geom_tiplab(), node=21)
geom图层美化分枝
知道了我们需要美化的分枝的node值,就可以进行美化
tree <- groupClade(tree, .node=44)
ggtree(tree, aes(color=group, linetype=group))
我们也可规定多个节点
tree <- groupClade(tree, .node=c(44, 47))
ggtree(tree, aes(color=group, linetype=group)) + geom_tiplab(aes(subset=(group==2)))
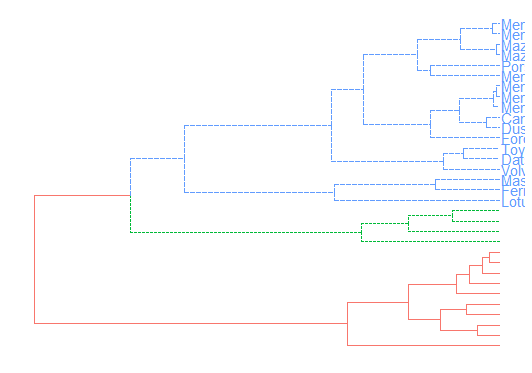
groupOTU美化端点
刚刚是关于分枝点(node)的美化,其实也有参数是进行树图最末端的端点进行美化的。端点就比较好理解,就是我们的样本名称。
tree <- groupOTU(tree, .node=c("D", "E", "F", "G"))#DDFG是样本名称
ggtree(tree, aes(color=group)) + geom_tiplab()

高亮分枝
geom_hilight对关注的区域进行高亮显示,scaleClade对关注区域进行扩大或者缩小。
ggtree(tree) %>% scaleClade(44, scale=0.3) + geom_hilight(44, "steelblue")
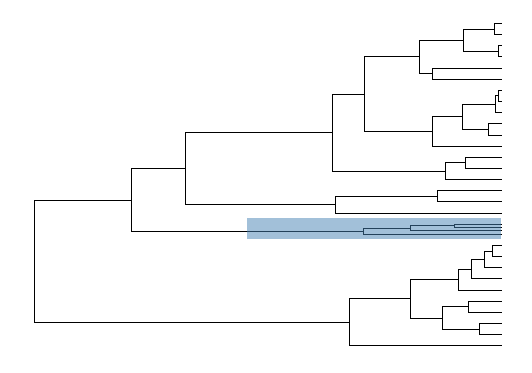
其他参数
用rotate旋转树图,用flip函数翻转两个选定的分支。

到此,我也算完成对ggtree的初步了解。完结撒花。