前言:这篇文章是我打字最多的了,而且对于网络图背后的图论知识,也是初涉皮毛。所以准备起来也是到处找资料。文中的观点不一定正确。但是也是目前我认为的正确。
[TOC]
1 背景
最近觉得随着生物学的发展,越来越多的场景需要用到网络的概念。主要是目前生物研究已经到了大数据的时代。这可能其中有两部分原因,一方面是随着生物学基础理论研究的不断深入。明星分子已经研究的差不多,目前可能主要是在研究信号传导网络,而生物体内的信号传导网络是非常复杂的。二是随着科技的发展和高通量组学实验的兴起,越来越多组学数据被挖掘出来。
这个时候,网络分析工具就显得十分必要。网络分析提供了一种灵活的分析工具。生物网络是生物学数据的直观可视化,因为它们可以同时处理大规模和多样性(scale and diversity)的数据。除了数据可视化之外,网络的一个主要优点是它们能够展示涌现性(emergent properties)。涌现性是那些如果网络的一部分被单独研究就不会被观察到的特性。这些特性可能有助于解释复杂系统的行为,例如它们明显的鲁棒性[1]或模块化(robustness or modularity)。
网络图的元素主要包括了点和线,点的话最好理解,就是我们要研究或者关注的目标,这个目标可以是一个蛋白,基因,代谢物,或者一个微生物菌种,甚至一个理化因子,
但是线的属性就比较复杂,线也是链接我们所关注目标之前的作用关系。这个作用关系首先第一要素是相互有所联系。但是这个联系的方式确实非常难以获取的。目前常用的是根据表达量取去计算相关性。不论是person,spearman,或者WGCNA中TOM值。除此以外,还有一些专门的计算网络节点之间关系的软件。但是我自己是将其分为两种。
- 节点之间有已知关系,比如string数据库,和KEGG数据库
- 节点之间没有已知关系,但是节点有交集的属性,比如基因与另一个基因的表达量属性。
[1] 鲁棒性:是指控制系统在一定(结构,大小)的参数扰动下,维持其它某些性能的特性。
2 网络图的基本概念
通常,网络具有某些属性,可以通过计算这些属性来分析网络的属性和特征。这些网络属性的行为通常定义网络模型,并可用于分析某些模型如何相互对比。
网路属性包括:大小 (size)、密度(density) 、平面网络密度(planar network density) 、平均度(average degree) 、平均最短路径长度(average shortest path length) 、 聚类系数(clustering coefficient) 、连通性(connectedness) 、节点中心性(node centrality) 、节点影响(node influence)等。详情见WIKI百科

因为网络科学是非常复杂的一个科学(至少对我是的),所以对于其中的一些具体的含义就不再过度的追究。这里就是复制一下帮助文档可以帮助我们在有需要的时候来进行查看。
我们主要讲cytoscape能计算出来的这些参数,毕竟是软件认证过比较重要的参数。
2.1 基础知识(可以跳过)
对于图论我也是刚刚了解,知道的也不多,只是目前先把自己的理解写下来,错误的后面再修正。
2.1.1 Size (网络规模)
网络规模由网络中所含元素的数量来衡量,比如某个网络中含有500个基因。
2.1.2 Network density (网络密度)
网络密度即网络连线的稠密程度,高密度提示网络存在更多的连线, 即词与词之间的连接关系更丰富。如果网络中的任意两个词之间均存在连线, 整个网络就处于完全连接状态, 其密度为最高值为 1。如果整个网络只有点而没有连线, 则其密度为最低值为 0。
2.1.3 Degree centralization (点度中心势)
点度中心势表示的是整个网络的集权化(cent ralize)程度, 反映了词的势力(以点度衡量)在整个网路中的集中程度, 以百分率表示。如果某个网络有一个词位于中心, 其他词均只与该中心词相连, 那么将组成一个完全“中央集权”的星状网络, 其点度中心势为最高值100 %。如果某个网络中的词首尾相连组成一个圆环, 那么所有点度均相等, 其点度中心势为最低值0 %。因此, 点度中心势越大, 网络连线分布就越不均衡, 最有势力(点度最大)的词与其他词的势力之间的落差就越大;点度中心势越小, 网络中的各个词的地位就越均等。
2.1.4 Number of connected components(中心点)
就是这个网络中有几个中心,数据越少代表网络内联通性强。
2.1.5 shortest path length (最短路径)
这其中有两个概念,一个是shortest paths,一个是 characteristic shortest path length。网络图中的路径是指两点之间,边的数目。假设一点为a,一点为b,从a到b最短有几个边,可以看到假设a直接和b相连,那最短的路径就是1。而特征最短路径(characteristic shortest path length)是对这个网络中节点之间最短路径的期望值。
2.1.6 Clustering coefficients(聚类系数)
聚集系数是表示一个图形中节点聚集程度的系数,证据显示,在现实中的网络中,尤其是在特定的网络中,由于相对高密度连接点的关系,节点总是趋向于建立一组严密的组织关系。在现实世界的网络,这种可能性往往比两个节点之间随机设立了一个连接的平均概率更大。
聚类系数是比率N / M,其中N是n的邻居之间的边缘的数量,并且M是n的邻居之间可能存在的边缘的最大数量。节点的聚类系数始终是0到1之间的数字。这个是最常用的。也就代表着,如果聚类系数越多,那么这个点(基因)可能链接非常多的点(基因)。很可能就是hub基因。平均聚类系数分布给出具有k个邻居的所有节点n的聚类系数的平均值。Network Analyzer还计算网络聚类系数,该系数是网络中所有节点的聚类系数的平均值。
2.1.7 Topological coefficients
拓扑系数,拓扑系数是节点与其他节点共享邻居的程度。Network Analyzer计算网络中具有多个邻居的所有节点的拓扑系数。拓扑系数越高,代表网络中存在越多的hub基因。
以上网络参数的分析可以用cytoscape计算,也可以使用pajek,Network X。
3 组学中的网络分析(可直接看这里)
BB了那么多,现在来点实际的。
3.1 趋势分析中网络分析
其实趋势分析并不是按照基因的表达模式聚为一类,而是类似WGCNA,根据基因的表达量计算之间相关性,再将相似的基因聚在一起,将代表绝大部分基因的表达模式认为是整体的特征。
虽然每个公司用的软件可能不同,因为 STEM (点击进入文献地址),是一个 java 软件,可能跑流程的话不太方便。但是基本原理都是 STEM 的原理。
STEM 这个软件的基本原理:
- 软件先按照预先的设定,模拟出n种最具有代表性的可能趋势(一种趋势就是一种基因表达模式);
- 计算每一个基因与预设的这些趋势的相关系数,然后将每一个基因归类到与其最相似的趋势中。
以上的两个步骤,也是STEM软件的1个特点:先预设趋势,再分配。这样处理的优点就是趋势更加有规律、整齐划一,便于后期解读。而其他大部分不预设趋势的方法(例如K-means聚类,虽然这个软件也有k -mean算法),聚类效果受数据的影响很大。在样本的时间点较少的情况下(3~5个时间点),STEM的聚类效果明显优于其他的聚类方法。当然,如果时间点太多的情况下(6个或6个以上),采用STEM去模拟所有可能的趋势,则会导致趋势过于零碎而加大后期数据整理的工作量,这个时候可以考虑使用其他的聚类算法。
参考网址:https://www.cnblogs.com/nkwy2012/p/7505079.html
那么如上呢,我们其实只是简单说了一下趋势分析的原理,但是其中涉及的 “ 网络参数 ” 并没有去涉及。这其中就有一个问题,如果老师想从目标趋势中进一步筛选候选基因集又该如何去做呢?
之前,我们一直推荐老师从
- 从KEGG代谢通路入手,关注那些和我们研究相关的重点的代谢通路进行研究。
- 从已知的明星分子入手,关注那些别人已经研究过的,或者在其他物种已经有证明在某些代谢通路具有非常重要作用的基因或者基因产物。
但是,这只是针对那些已经有比较明确目标的老师。如果老师没有明确的目标又该如何让老师去进一步筛选候选基因呢。最近呢,有一北京的老师就发来一篇文献。

文中其中就是从网络图的算法上入手,寻找其中的hub gene。作者用了其中三个参数。
- Clustering Coefficient ( 聚类系数 )
- Degree ( 连通性 )
- k-core ( k 中心值 )
首先聚类系数比较和联通性都比较好理解。degree是衡量某个基因与所有与它相关的基因之间关系大小。越大代表这个基因的 “ 人缘 ”越好,与它有关的基因越多。聚类系数也是类似的含义,也是衡量某个基因与周围基因的相关性。越大代表这个基因位于核心地位。这篇文献的英文说明,我觉得还是不错的。分享一下,大家一起学习一下。
Degrees describe the number of single gene that regulates other genes represent the size of the cycle node. The higher the degree, the more central the gene occurs within the network. The clustering coefficient can be used to estimate the complexity of interactions among genes that neighbor the core gene with the exception of core gene participation. The lower the clustering coefficients, the more independent of the core gene are the interactions among genes in the neighborhood of the core gene. A k-core of a gene co-expression network usually contains cohesive groups of genes. The higher the k-core, the more central the genes occurs within the network.
但是其中这个k-core其实我就不理解了,根据STEM这个软件的文献报道中,所阐述的那样,用的聚类方法是不同于那些主流的聚类算法 ( 层次聚类,*k-*均值聚类,自组织映射)。而是采用了一种新型的聚类算法。因为作者认为传统的主流算法都忽略了连续时间点之间的时间依赖性。具体来说,如果我们随机排列时间点的顺序,这些方法的结果不会改变。这很显然在生物学研究中是不合理的。
并且STEM这个文献,通篇都没有提到 k- core 这个参数。而 k- core 参数其实只是对 k -mean 聚类算法的一种矫正。因为 k -mean 算法 首先是
- 对原始数据,初始随机给定K个簇中心。可以想象成目前有 5 w 个基因,先随机给10个中心,这个时候给的簇中心点是随机给的。当然是不准确的。
- 然后计算各个基因之间的距离,这个里的距离算法很多,比如我们常见的欧式距离,曼哈顿距离等等,这个时候,我们自然就得知了基因之间的距离大小。之前我们假设了 10 个中心。自然我们就可以知道每一个基因到这 10 个中心点的聚类。将基因划分为最近的中心点,这样自然就形成了簇。
- 可以看到上面我们聚成的簇,其依据是我们随机选的,接下来就是用到计算机强大的循环计算进行迭代。如何迭代呢,就是计算上述步骤得到的每个簇的平均值点,重新认为是新的中心点。重新进行 2 步骤,这样不断的迭代。
- 直到聚类中心不再进行大范围移动或者聚类次数达到要求为止(比如我们规定 10 次)。
那么了解了k -mean 算法,那么 k-core 也是非常类似的。只是为减轻K-means算法对孤立点的敏感性,k-core 不采用簇中对象的平均值作为簇中心,而选用簇中离平均值最近的对象作为簇中心。但是自始至终我都没有发现k- core聚类算法中有这个参数的出现。而且看起来这个参数还是每一个点(基因)都有的参数。不太像构建网路时的整体参数设置。
反正STEM这个软件我猜测是不会给这个参数的。
参考资料:
那么只好继续找资料了,那么就发现:
分析表征网络的中心核理论。根据经典网络中心核理论, “ 中心核 ” 是位于整个网络结构中的子结构(子网络), 其由多个要素 ( 点 ) 构成, 在变动的环境中具有相对稳定性。常用的是k- core decomposition 这个方法去发掘网络中的稳定结构。 k-core 法有点类似于 “ 剥洋葱 ” 的方法:
- 从网络中提取满足以下条件的子网络。
- 该子网络中的每个节点至少与 k 个其他节点相连。
- 不满足 2 条件的节点都会被抛弃。
- 这样剩下的网络结构中每个节点都是满足 2 的要求。
此 k -core 的值非彼 k- core聚类算法!!!
这里的 k -core 的值 (老师发来的文献中的值)其实更应该叫 k 值,就是这个节点与其他节点相邻近的个数(我猜的,但这个不重要反正软件都会算的)。一个节点的特征性k 核值越大, 该节点就越不容易被删除, 而且该节点所处的子网络结构也越不容易受损, 因此就可以得到从周边到核心的网络结构的分层解析。
参考文献:
- 教学对社会表征的塑造:对词汇网络的研究
- Genes related to the very early stage of ConA-induced fulminant hepatitis: a gene-chip-based study in a mouse model
3.2 WGCNA 中的网络分析
3.2.1 WGCNA分析概述
3.2.1.1 WGCNA的名称
正如我们之前所说的,《网络图的理论基础初探(一)》,网络分析越来越多地用于生物信息学应用。其中最著名的怕是今天要说的 WGCNA 分析,也就是我们常说的基因权重共表达网络分析。
注意,WGCNA的名称有重点:
权重,这有两个含义,
- 一个是那就必然涉及到某种数据上的处理,也就是说这一种根据基因的某种特性而作为标准来分析的 “加权” 分析的方法。
- 以及 WGCNA 是一个权重分析网络(是否有硬阈值和软阈值之分)。这个概念可能比较难理解。这里我们可以套用 WGCNA 的作者的话。英文。我就不粘贴了,因为我的 markdown 水平还不会制作公式。很尴尬。我就简单说一下。非权重网络也是我们生物学分析常用的网络方法,其和权重网络的区别就在于采用的阈值的区别。
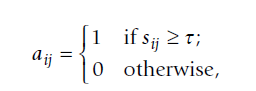
只有大于 那个我不会打的字 的相关性才会被认为有,低于就认为无。这很显然是不合理的,所以作者才进一步的用非权重网络做 WGCNA 分析。也就是说,在计算 i 与 j 之间的相关性后,会用一个系数加权。(本意就是全部的相关性都会做考虑)
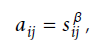
共,既然是共,那自然是将基因放在一起(模块)做分析啦
表达,至少要有表达量或者类似表达量这样的连续性变量吧
3.2.1.2 WGCNA的介绍
WGCNA 分析是个 R 包,是08年发表的在 《BMC Bioinformatics》的一个生信分析 R 包,WGCNA分析有多火呢。大家可以看一下引用率(截至19/1)。
可以说随着测序价格的不断降低,以及组学研究的不断深入,目前很多老师的项目样品数都是非常多的。基本上只要实验设计中涉及到时间点的影响或者两个以上因素的实验设计的样品数都是可以达到WGCNA分析的样品数最低要求。
| 实验设计 | 样品数 |
|---|---|
| 有生物学重复的 | >= 8 |
| 无生物学重复的 | >= 18 |
WGCNA R软件包是R函数的综合集合,可以实现的WGCNA的各个分析。该软件包包括网络构建,模块检测,基因选择,拓扑属性计算,数据模拟,可视化以及与外部软件接口的功能。
但是 R 包的教程不是今天要说的重点,我也不会,尴尬,不过WGCNA分析跑完整个流程是不难的,为什么我这么说呢,因为网上教程太多了。0.o,这个理由够不够充分呢。
所以想自己分析的,可以跳转一下链接。
官方英文版:
https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/index.html
中文版:
http://www.bioinfo-scrounger.com/archives/260
另外附一个小的 R 代码,因为 WGCNA 包是放在 bioconductor 上的,设置 setRepsitories 参数就可以用install.packages 直接搜索安装。美滋滋。
setRepsitories(ind = 1:2)
install.packages("")
3.2.1.3 WGCNA 分析的目的和流程
其实目的嘛,最直白就是为文章提高格调啊。0.o
当然 WGCNA 分析可以帮助我们缩小候选基因集,很多时候,老师做完转录组,甚至全转录组,数据一大堆,根本不知道该如何分析。这个时候往往就需要结合各式各样的分析方法,从中挑选一些我们能写文章,能讲故事的数据。
WGCNA 就是这样一个分析方法,让我们将数据进行处理,筛选掉不关注的,不重要的基因,留下我们关注的基因。一句话,缩小范围!
这也是 WGCNA 分析最核心的功能吧,至于软件作者说的寻找 biomarker,寻找核心基因,关键基因模块什么的,巴拉巴拉都是官话。
至于流程嘛,网上说法蛮多的,我自己稍微总结下吧
- 过滤数据,对,就是先过滤数据
- 数据处理,符合无尺度网络
- 划分模块
- 以模块为单位,进行数据分析
- 模块内,外的网络分析
- 挖掘 hub gene 或者调控网络(出图)
3.2.2 WGCNA 分析流程
3.2.2.1 过滤数据
过滤数据之前,当然要先有输入的数据,WGCNA 分析中,输入的数据有两个矩阵,也叫数据框吧。
- 表达谱数据,Notice:表达谱不仅仅只是说转录组!!!是指任何有表达量或者连续变量数据的分类变量。(自行体会,发现自己自从看了 《R for data science》,说话越来越绕口了 )
- 样品性状的数据,也就是表型数据(大部分项目都没有提供这个数据)
当然,表型数据不是必须的,只是没办法做其中模块与表型的关联分析罢了。
为什么要过滤数据呢,首先基因太多,影响分析速度,其次背景噪音多,影响分析的精度。最后基因太多,增加后续分析难度。
如何过滤呢
- 去掉低丰度的基因。(自己设置阈值)
- 去掉变化不大的基因。(变异系数是不错的标准)
3.2.2.2 数据处理
生物学中,复杂的网络信号传导调控机制并不是随机发生链接的,而是存在一些核心基因,也就是一些转录因子,蛋白激酶等等,这些核心基因位于信号传导途径的中心地位,响应上游信号,并调节多个下游的信号途径。
而传统的网络分析都是忽略了这一点,而 WGCNA 分析通过将基因之间的相关性进行了 n次幂的取值。
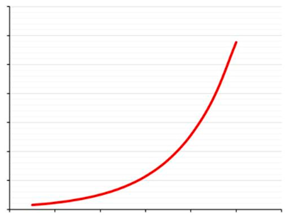
也就是说进一步的扩大贫富差距,这样就造成了富者越富,穷者越穷。相关性高(比如0.95)的基本不受影响,而相关性低的(比如0.5)就很快就变得非常低。
但是这样得处理,恰恰反应了生物学信号网络的特性,就是大部分基因都是执行者,小范围的,本本分分的执行上级安排的任务。少部分基因具有强大的影响力,调控多个下游基因。整体上影响信号网络。比如 wus, STM,p53等基因。
这样的网络也叫无尺度网络:
无尺度网络:满足上文提到的无尺度网络的定义假设m为某节点的连接数。统计所有基因的m值,然后以m高低为指标对所有基因分类。n为节点连接数为m的基因的数量。m与n应该成反比
原始矩阵:Smn=|cor (m, n)|#注意这里是绝对值,也就是说WGCNA分析只考虑强弱,不会考虑正相关还是负相关
无尺度化(拉大贫富差距),确定最佳β值。 aij=power (Smn, β)=|Smn|^β
所以 WGCNA 首先第一个参数就是 n次幂(β)到底取什么值
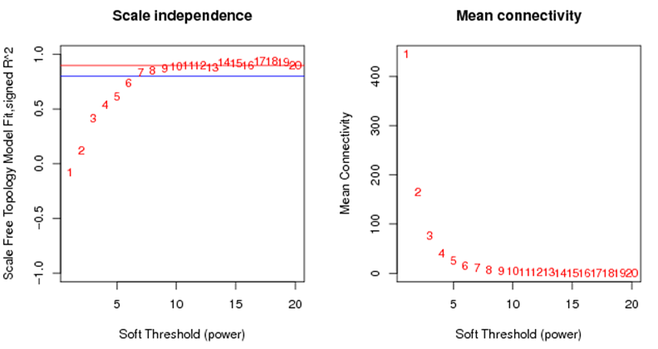
WGCNA 分析中都会有这样的图。左图:不同β值下,m与n的相关性的变化。就是我们刚刚说的无尺度网络分析的定义。一般认为取β值大于0.8或到达平台期时最小的β值用于构建网络。右图:不同β值下,所有基因连通性的均值。
3.2.2.3 划分模块
划分模块,有点类似于我们在用 pheatmap 包中的 cut_tree函数,但 WGCNA 的方式更为高级一些。是动态剪切树算法(dynamic tree cutting)分割模块。原理可以看可以看这个博文。
首先呢,WGCNA 分析计算基因与基因之间的 TOM 值,TOM值就很厉害了,它可以计算间接关系。但这个是另一个故事了,就不再多说了。(主要是我写不动了,早点结束WGCNA就好了)
但是,我们只需要了解 TOM 值可以计算两个基因的直接关系以及间接关系。简单理解为相关性就好。
知道了基因间的相关性,就可以绘制热图。最关键的是可以根据这个热图上的信息,计算距离绘制聚类树。再使用切割树的算法,将相关性强的基因聚在一个模块内。
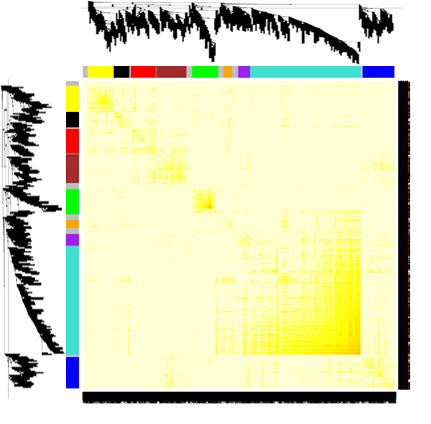
这里有个小 tips ,TOM值是划分模块的重要参数,同时也是衡量基因与基因之间相关性强弱的指标。
3.2.2.4 寻找模块
做 WGCNA 最关心的目标主要用找到关键的基因集合
找到关键的基因集合,这个范畴就很广阔了,但是比较主流的是用生理生化、环境因子,处理效应,SNP,临床数据,其他组学数据,等等都可以作为一个外部的性状,进行性状关联分析。找到我们相应比较关注的模块。这里的相关性分析也是利用外来的一个标度去衡量我们的模块到底那些和我们关系的问题相关。但是目前由于老师了解的少,我也接触也很少,目前还没做过任何一个做性状关联的 WGCNA !!这是因为很多老师的实验设计中,样本本身就带着一个 “ 某个维度的分类 ”,也就是样本本身就是带着性状的信息的。所以很多老师都是看着自己的样本与模块之间的相关性来选择的。
但是,如果我有性状,那 WGNCA 又是如何分析。首先 WGCNA 会计算 Gene significance 值,基因显著性(GS)。数学公式 又要从头说起了,平时我们用 R 计算矩阵的相关性。
虽然就一个 cor 函数就行了,但其实其中发生的计算还是很复杂的,简单来讲就是计算列于行之间的相关性。假设列是样本,行是基因。我们就可以计算基因与样本的关系。但是我们是可以增加一列,比如我有一组临床数据,这个时候就可以加入一行性状的数据,从而去描述变量。假设性状数据T, T=(T1, . . ., Tm)已知,就可以依照下面公式算出 Xi 基因与性状数据之间的相关性。显而易见,这个相关性 GSi 越大,这个基因与性状之间的关系越强。
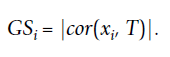
但是呢,很明显 WGCNA 之所以花了那么多时间分模块,如果直接计算每个基因与形状之间的相关性,挑出最大的不就行了。
这是不行的,因为数据量远比我们想象的大,生物问题之间有没有很好的划清界限,所以划分模块是最快最有效的即考虑生物学问题,又能从庞大的数据快速抽出一个小范围研究对象的方法,也就是我们常说的缩小候选基因集合。
不论是我们根据性状关联分析的结果,还是根据根据模块与样本间的相关性,甚至我们只关注基因的表达模式,把 WGCNA 当成最简单的多样本的趋势分析。然后根据模块内的基因表达模式来选择模块(注意那个图表达模式的图的纵坐标是模块特征值Module eigengene E)。而不是表达量奥,这就意味着不是所有模块的基因都是符合这个表达量变化的趋势。因为只是一个模块特征值,有点类似 PCA 分析中 PC1!!!
趋势分析也有这个毛病奥。看上文,帮助理解。
总之,我们挑选出来一个或者数个我们关注的模块。
那么接下来就是 模块内的分析了。
3.2.2.4 模块分析
模块分析的方法很多,大概分为两种
- 系统性总览模块特征
- 局部性深入探索数据
一,系统性总览模块特征
(1)第一个要说的是模块内基因热图,就是相同模块内的基因相关性会大一点,这个图我感觉吧,就是为了说明分模块分的好。数据挖掘的意义不大。
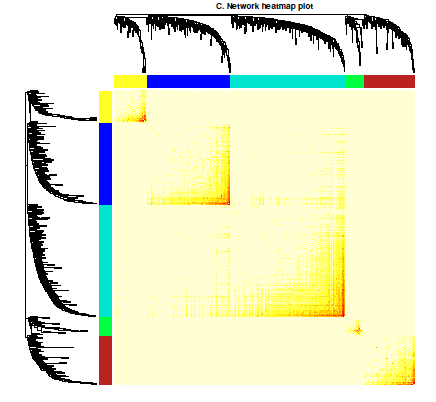
热图的意思就那样,其他的类似的也懒得解释了,各位有结果的时候自己看吧
(2)网络性分析
网络之所以成为网络是有自己的特性的。比如我们之前说的网络图论中那些名词解释,都是对网络概念非常重要的点。
下图就是通过计算k值, 也就是整体网络的连通性之间与的corresponding frequency(响应频数)比值来展现网络图的网络特性。好吧,响应频数又到了我的盲区了。各位知道有这么个东西就行了。那天有空再搜索一下。
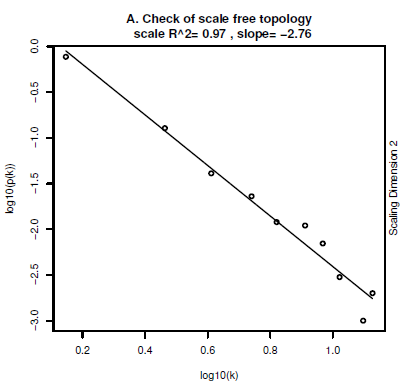
这个图就是反应了如果两者的比值是一个近似直线,就代表这是一个自由的拓扑网络。我觉的意思就是这是个好的网络呗。纯纯的网络,不含一点杂质,黄金奶源,不是所有牛奶都叫特仑苏。
二,局部性深入探索数据
数据探索,花样就多了。
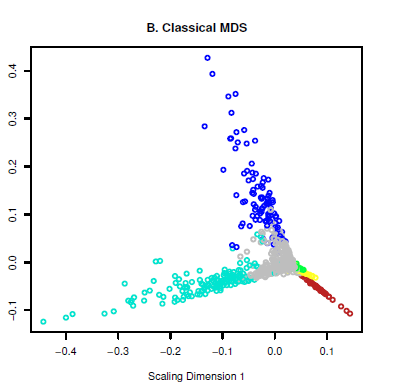
(1)选一个模块内的基因做个MDS分析,越靠近手指的部分的基因越是hub gene
(2) 做了性状关联分析就知道了每个基因的GS值,那么一个模块内的所有基因的GS的平均值也可以反应这个模块与性状之间的关系,如下图
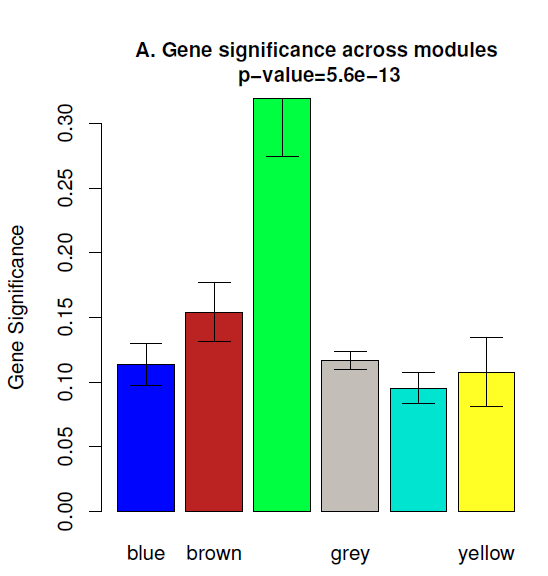
可以看到上图是绿色的柱子的GS总值最高。也就是说目前green这个模块可能最相关。但是 WGCNA分析中还有一个概念叫做 Module membership (MM)也叫做Kme值。
是衡量基因和模块之间的相关性的,这个值之前老师问过我,这也是网络图系列公众号文章的写作初衷,就是加深自己对WGCNA 分析的了解。
不想细说,具体看还没出品的《网络图(四)常用网络分析中的概念与名字解释》
总之,MM越大,说明这个基因与这个模块的关联性越大。
那么我们刚刚知道了green模块有意思,那其中那个基因最有意思呢,可以利用MM一探究竟嘛。
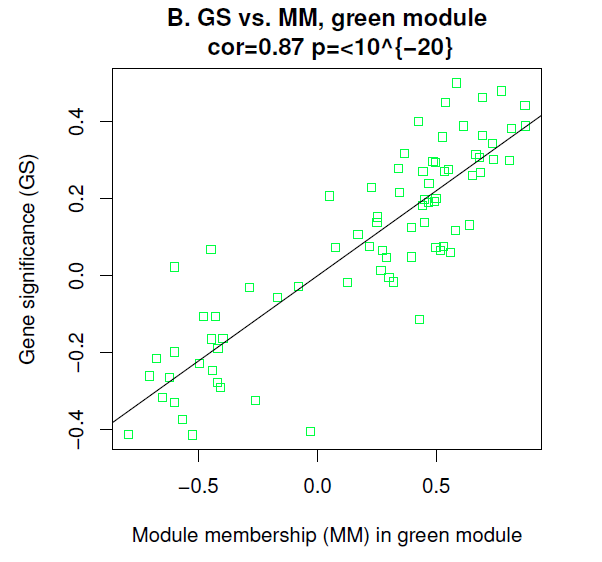
很有意思的是,我们额外发现,MM值越大,GS值也越大,这更加说明了这个模块很有意思,和性状很相关。
3.2.2.5 关键基因
关键基因的的方法就是
- 功能注释,富集分析
- 网络分析—connectivity等值筛选
第一个功能和富集分析就不说了,玩烂了
Hubgene 是我们认为的网络调控关键基因,往往是蛋白激酶,转录因子等等,这种位于上游的调控因子往往是无尺度网络中的核心,也就类比于,铁路线中的郑州,北京等大站。所以很多基因都是和 Hubgene 有联系的。不管间接还是直接作用(考虑间接作用是WGCNA分析的大亮点)。所以 WGCNA 的作者也说了,Hubgene 是高连通性的gene(genes inside co-expression modules tend to have high connectivity.)所以connectivity是WGCNA作者推荐的,就别想其他了。
GS值,上文讨论过了。可以试试的
Kme 不推荐,kme 值也叫Module Membership,顾名思义就是模块关系,是基因与模块之间的关系。如何计算基因与模块之间的关系,就是计算基因的表达量与模块特征值之间的相关性。其实Kme值 只是衡量基因与模块之间的相关性。绝对值越大,越接近1代表这个基因和这个模块越相关。所以会被认为是这个模块内的。但是这个参数也只能衡量基因与模块之间的关系。和 hubgene 的概念并没有什么直接的交集。
3.2.3 网络图的可视化
可视化还是非常重要的,常用的就是 cytoscape 了。windows就可以下载安装,可视化图形界面,老师的首选。
cytoscape软件的使用:
cytoscape是常用的网络图绘制软件,老师后续有需求可以自定义网络图的形状,线条,颜色等。
https://cytoscape.org/download.html (需要先安装java或者jre,百度jre或者java,下载即可(https://www.java.com/en/download/win10.jsp)
安装后,点击cys格式的文件,并可以在操作界面中自定义图片。
cytoscape课程:http://www.omicshare.com/class/home/index/series?id=3
