最近很长时间都没写公众号了,倒不是因为工作忙,虽然这周事情确实多。不多主要原因是最近,沉迷10x单细胞,自己跑了单细胞分析的几个R包,确实能加深对产品的了解。此外,仅有的时间也在研究差异分析DESeq2。
前言
最近很长时间都没写公众号了,倒不是因为工作忙,虽然这周事情确实多。不多主要原因是最近,沉迷10x单细胞,自己跑了单细胞分析的几个R包,确实能加深对产品的了解。此外,仅有的时间也在研究差异分析DESeq2。
其实这两个事情是两个极端,基本上各个测序公司和生信公众号都在推送关于单细胞测序的相关内容。以往我们研究组学,都是在宏观的bulk样品上去研究基因和基因产物。但是10x单细胞技术,给大众带来了从单个细胞的水平上去研究生命的的可能性。虽然生物学实验技术发展到今天,各式各样的方法都很多。但是真正能分选出单个细胞。并且获得丰富的基因丰度和变异信息的。也就是17年来发展迅速的单细胞测序技术。
10x这是个新技术
但是DESeq2进行差异分析,却不是什么新鲜的东西。从09年的edgeR到后来的DESeq,差异分析软件可以算是伴随高通量测序发展至今的老技术了。但是老不代表重要,恰恰相反,最早开始的技术才是高通量测序中最重要的。我们进行高通量测序,获得两个维度的信息,一个是序列信息,一个是丰度信息。
有了丰度,就要比较丰度的差异,就要探究处理之间的差异。控制变量,寻找差异,是任何实验亘古不变的主旨。
最近17年刚刚发表的DESeq2对旧版本的DESeq进行优化和改进。而edgeR却没有任何动静,不少分析人员都转身到DESeq2或者EBSeq的怀抱中去了。
但是edgeR可以分析没有生物学重复的奥,甚至edgeR还想出了四种方法来矫正没有生物学重复。话说,我觉得做实验不设置生物学重复的都是耍流氓。但是edgeR确实能处理这样的疑难杂症,对于我这种售后技术支撑来讲还是很NICE的软件,但是各位老师,生物学重复还是不能不设置的。切记切记。
此外,DESeq2和edgeR的分析中,都各自有一套矫正。简单说就是
- 先过滤低丰度基因,
- 选择参考样本,
- 计算矫正因子,进行矫正数据
- 根据基因的表达情况过滤
- 差异分析,计算p值,矫正p值
但是这里面每一步edgeR和DESeq2计算都不一样。其实两个计算方法都差不多,但我个人更喜欢DESeq,为什么因为它新,它后出,它更新了。对于一些edgeR的不周全之处,它会考虑。差异具体最重要的三点就是:
DESeq2取log值,降低异常值的影响- 过滤基因是根据平均值大于阈值来过滤(但同时异常值不会被考虑),而非
edgeR在两个样本上表达。(其实这个感觉因人而异) - 而上述的平均值筛选的阈值,是根据计算得来的,是软阈值,优势不言而喻。
当然其中还有大量的,我不了解的参数和计算过程,毕竟我也是很初步的了解两者。
但是今天这篇文章主要还是讲如何利用DESeq2进行差异分析。
以及后续我会根据自己的理解来整理一下edgeR于DESeq2的原理
以及后续整理一下DESeq2内在的一些细节上的参数(这也是我学DESeq2的根本原因,了解什么参数如何调整从而优化结果)
DESeq分析
我只是个技术,不是生信分析,所以我不会linux,也不会python和perl,唯一还算有点了解的还是R,幸好DESeq2和edgeR都是R包,要不然我是不会去敲代码去了解的。
输入文件
了解生信分析软件,最关键的是输入文件和输出文件。
DESeq2的输入文件有两个,一个是count值计数文件,count值就是比对上这个基因的reads数目。一般公司给的结果都有的,这次我也是用公司给的表格数据来做的。因为我不会linux,自然也不会hisat2等比对软件咯。所以上游是断,只有从已有的公司结果出发。不过我相信大部分公司都会给count值的。比如我所在的公司就是在表达量总表里。
DESeq2需要的输入文件是这样的,行为基因名称,列为样本,值为count值
## untreated1 untreated2 untreated3 treated1 treated2
## FBgn0000003 0 0 0 0 0
## FBgn0000008 92 161 76 70 140
## .....
另一个就是样本设计的表格,也就是分组信息,当然也可以多个分组,从而计算批次效应。
## condition type
## treated1fb treated single-read
## treated2fb treated paired-end
## treated3fb treated paired-end
## untreated1fb untreated single-read
## untreated2fb untreated single-read
## untreated3fb untreated paired-end
## untreated4fb untreated paired-end
表格整理
知道了输入文件的格式,就努力整理数据表格吧。
相信了解的R的人,处理起来是不难的,要是实在不行,就用excel
我的命令如下,其实也是半自动化的,理论上,只要count值那一列的名词里有“count”,行是基因名词的数据,应该都可以的。有兴趣的话,可以用自己的数据试试,如果出错,很可能是正则表达式的错误用法。
# R 3.5.2 win 10
library("`DESeq2`",quietly = TRUE)
library(magrittr)
library(stringr)
#browseVignettes("`DESeq2`"),这是官方说明文档,不看说明文档的生信分析就是耍流氓
#处理原始数据,要看公司给的那个表格中有count值的信息
#把infile设置为这个包含count值的表格,复制到工作目录下
infile <- "all.genes.expression.xls"#读取的count数文件
path <- "C:/Users/woney/Desktop"#这是我的桌面路径,注意windows自带的反斜杠和R认为的反过来的
setwd(path)#设置工作路径
expstat = readr::read_tsv(infile, col_names = T) %>% tibble::column_to_rownames(var = "id")
#用readr的包是因为read.table老是出各种叉子
sample <- colnames(expstat)[stringr::str_detect(colnames(expstat),"count")]#这里用str_subset()会更好,但我懒得改了
#读取count数据
count <- expstat[,sample]#只读取表达量总表中的有count值的列
count = round(count,0)#`DESeq2`只能处理整数的count值
当然我不能保证上面的命令,粘贴复制就管用,所以学习R,弄懂每一行命令的含义最重要。
上面我们已经读取了count值得数据,下面就要设置样品分组的colData数据了
#设置分组colData的信息
sample <- stringr::str_replace_all(sample, "_count","")
colnames(count) <- sample
group <- stringr::str_replace_all(sample,"-.*","")
condition <- as.factor(group)#一定要是因子类型的向量,切记
colData = data.frame(row.names=sample,condition)
那么设置好输入文件后,就要进入差异分析了。但是差异分析前,要设置好参数
参数设置
diff_set <- "PBS_VS_GFP" #设置比较,后面比前面,PBS是对照
diff <- unlist(str_split(diff_set,"_VS_"))
cutfc <- 1 #差异倍数的阈值,默认是是差异两倍,也就是log2的1
test <- "FDR" # "PValue"根据p值筛选,就换成test <- "PValue"
cutp <- 0.05 #统计检验值,小于0.05认为统计上显著
差异分析
我这里只是列举了简单的差异分析,不列举参数,因为我还没了解明白
## 差异分析
dds <- DESeqDataSetFromMatrix(count, colData, design= ~ condition)
dds$condition <- relevel(dds$condition, ref = diff[1])
dds <- DESeq(dds)
#以下都是差异分析中的步骤,列举出来有助于我们了解其内在的分析流程
#estimating size factors
#estimating dispersions
#gene-wise dispersion estimates
#mean-dispersion relationship
#final dispersion estimates
#fitting model and testing
res <- results(dds, contrast=c("condition",diff[2],diff[1]))
res就是差异分析的结果,但是大家输入res就会发现,DESeq2的输出结果中没有表达量的信息,但是有log2FC和P值、Q值的信息。一般来讲,做差异分析,肯定是想知道表达量的。然后再进行差异分析。但是其实这里面不是承上启下的流程。而是输入count值,出来差异倍数和检验值。虽然根据
diff_gene_`DESeq2` <-subset(res, padj < 0.05 & abs(log2FoldChange) > 1)
也能得到差异基因的列表,但是没有类似于FPKM等表达量信息,没有表达量就没有其他那些基于表达量的信息了啊。但是DESeq2就是没输出,我觉得可能是表达量的计算方法有很多,而edgeR和DESeq2都是根据CPM来计算的,因为CPM不矫正基因的长度,所以基因的表达量的绝对大小会有比较大的波动。这意味着有大量的信息可用于离散估计和差异测试。
同时说实话,大家都是比较同一个基因,都是一个基因,矫正长度有什么用,都是一样的长度。
所以说实话,这也是FPKM等对长度进行矫正的表达量公式地位下降的原因。
总之,我们需要表达量,那么就要计算表达量。不同样本间的同一个基因比较,跟长度关系不大,直接用CPM来计算,为什么呢,因为我没有基因长度信息。R语言处理DNA序列,天生就不强,再说我也不会。更不会用其他非R软件去计算。
CPM与FPKM的比较:https://support.bioconductor.org/p/69433/
表达量计算
#计算表达量
diff_sample <- str_subset(nms,pattern = paste0(diff[1],"|",diff[2]))
data = count[,diff_sample]
## cal cpm
data2=data
allcols <- length(diff_sample)
for(i in 1:allcols){
data2[,i] = round(data[,i]*1000000/sum(data[,i]),2)
colnames(data2)[i] = paste(colnames(data)[i],"_CPM",sep="")
}
## mean
C_number = length(diff[1])
data3=data2[,1:2]
data3[,1] = apply(data.frame(data2[,1:C_number]), 1, mean )
data3[,2] = apply(data.frame(data2[,(C_number+1):allcols]), 1, mean)
data3[which(data3[,1]==0),1]=0.001
data3[which(data3[,2]==0),2]=0.001
log2fc=round(log2(data3[,2]/data3[,1]),4)
colnames(data3)=paste(diff,"_mean",sep="")
## ouput file, id,count,log2(fc),pvalue,qvalue
data4=data.frame(rownames(data),data,data2,data3,check.names=FALSE)
colnames(data4)[1] = "id"
data4$'log2(fc)' = log2fc
data4$PValue = (res@listData)$pvalue
data4$PValue[is.na(data4$PValue)] = 1
data4$FDR = (res@listData)$padj
data4$FDR[is.na(data4$FDR)] = 1
这就有了表达量、count值,差异倍数,和检验值。
但如果你仔细看,就有疑虑,这个疑虑我现在不能说,看一下就好。
结果检验
因为我同时还有edgeR进行分析。以及根据DESeq2的结果直接进行分析。以及根据DESeq2和检验值,和我自己算的表达量都进行计算。
差异如下:
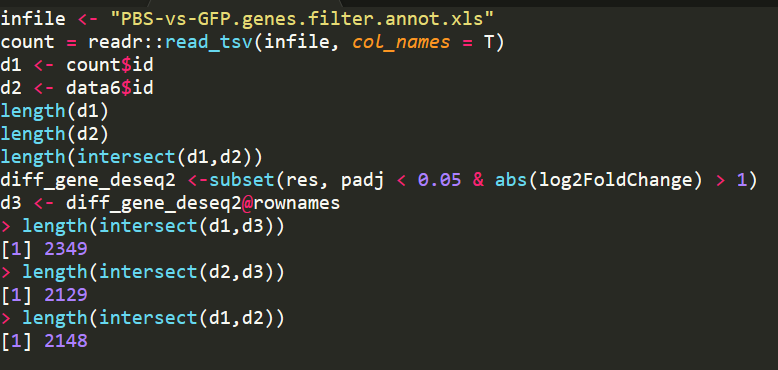
同样的差异分析阈值,FDR < 0.05 和 log2|FC| > 1,得到的差异基因数目如下:
忘了保存了,导致忘了具体的)
| edgeR | DESeq2 | myFC + DESeq2 |
|---|---|---|
| ~2500 | ~2400 | ~2300 |
交集在各自的图中,没有韦恩图可视化,因为我还在摸索韦恩图的R包
简而言之,我的感觉是